胆囊收缩素通过胆囊收缩素受体A减弱炎症反应降低脂多糖诱导的急性肺损伤*
2022-11-30廖勇杰岳嗣凤
谭 媛, 廖勇杰, 岳嗣凤
桂林医学院附属医院新生儿科,桂林 541001
急性肺损伤(acute lung injury,ALI)表现为胸部X线片显示双侧肺弥漫性浸润,动脉血氧分压与吸入血氧浓度比值(PaO2/FiO2)≤300 mmHg和肺动脉楔压(PAWP)≤18 mmHg或临床无左心房高压证据。根据PaO2/FiO2比值差异分为轻度、中度、重度急性呼吸窘迫综合征(acute respiratory distress syndrome,ARDS),PaO2/FiO2≤300 mmHg为轻度;PaO2/FiO2≤200 mmHg为中度;PaO2/FiO2≤100 mmHg为重度。ALI发生过程中,炎症紊乱导致肺内皮和上皮屏障破坏,其细胞层面变化表现为肺泡-毛细血管膜完整性丧失,中性粒细胞过度迁移并引起促炎因子释放[1]。研究发现,新生儿ARDS发展进程迅速,死亡率高,而早产儿由于肺部发育不完全,肺部更易受损[2-4],肺部感染引发炎症同样是新生儿ARDS的重要发病原因之一[2]。新生儿急性呼吸衰竭(包括ARDS、肺炎等)也常常伴有过度炎症反应,更有动物实验发现,抑制炎症反应有助于减轻新生幼仔肺损伤[5-6]。
胆囊收缩素(cholecystokinin,CCK)是一种多肽类激素,其受体有两种,包括胆囊收缩素受体A(cholecystokinin A receptor,CCKAR)和胆囊收缩素受体B(CCKBR)[7]。研究发现CCK能通过抑制炎症和细胞凋亡保护小鼠肝脏缺血再灌注损伤,同时其受体CCKAR表达上调[8]。但CCK与CCKAR是否参与脂多糖(lipopolysaccharide,LPS)诱导的ALI仍未见报道。近年来,随着生物科技的飞速发展,各种新兴手段如基因芯片、高通量筛选等被广泛应用于ALI发病分子机制研究[9]。本研究利用生物信息学手段预测发现CCKAR与LPS诱导的ALI相关,并探讨CCK及CCKAR对LPS诱导的炎症反应的影响。
1 材料与方法
1.1 材料与试剂
人正常肺上皮细胞BEAS-2B购自武汉普诺赛生命科技有限公司;支气管上皮细胞培养液(BEGM)Bullet Kit购自Lonza公司;CCK、LPS、MTT试剂及拮抗剂丙谷酰胺(Proglumide)均购自Sigma公司;兔抗人一抗CCKAR、phospho-p65(p-p65)、p65、GAPDH购自Abcam公司,兔抗人一抗phospho-IκB(p-IκB)、IκB和山羊抗兔二抗蛋白抗体购自Cell Signaling Technology公司;BCA试剂盒和RIPA蛋白裂解液购自南京碧云天生物技术公司;ECL发光检测试剂盒购自上海爱必信生物科技有限公司;人IL-1β、IL-6及TNF-α ELISA Kit购自R&D System公司;过表达质粒载体(pcDNA3.1)购自BioVector质粒载体菌种细胞基因保藏中心;Trizol试剂,SuperScriptTMⅣ第一链合成系统,PowerUpTMSYBRTMGreen预混液购自Thermo-Fisher Scientific公司;CCKAR引物:F5′-GACGC-TTCGGTCATTAGA-3′,R5′-GACGCTTCGGTC-ATTAGA-3′;β-actin引物:F5′-GGGACCTGACTGACTACCTC-3′,R5′-ACTCGTCATACTCCTG-CTTG-3′购自Invitrogen公司。
1.2 方法
1.2.1 细胞培养及实验分组 使用BEGM Bullet Kit培养BEAS-2B细胞,孵育箱培养环境为37℃、5% CO2。当细胞生长至约80%汇合度时,使用0.25%胰酶消化细胞。取对数生长期细胞进行后续实验。
将BEAS-2B细胞分为6组:未经任何处理的细胞为空白对照组(Control);利用LPS(1 mg/L)诱导BEAS-2B细胞24 h,并命名为LPS处理组(LPS);将过表达空载体转染至BEAS-2B细胞后,使用LPS(1 mg/L)处理24 h,并命名为CCKAR过表达阴性对照组(LPS+OE-NC);将CCKAR过表达载体转染至BEAS-2B细胞中,使用LPS(1 mg/L)处理24 h,并命名为CCKAR过表达组(LPS+OE-CCKAR);BEAS-2B细胞经CCK(10-6mol/L)处理1 h后,使用LPS(1 mg/L)进行24 h诱导,并命名为CCK过表达组(LPS+OE-CCK);BEAS-2B细胞经CCK(10-6mol/L)和Proglumide(25 μg/mL)共处理1 h后,使用LPS(1 mg/L)进行24 h诱导,并命名为CCK与CCK拮抗剂组(LPS+CCK+Proglumide)。
1.2.2 RT-PCR法检测基因表达水平 利用TRIzol裂解液提取细胞中的总RNA,依据说明书,使用SuperScriptTMⅣ第一链合成系统合成cDNA,使用PowerUpTMSYBRTMGreen检测CCKAR在不同分组中的相对表达水平。
1.2.3 Western blot检测蛋白表达水平 利用RIPA蛋白裂解液提取细胞中的总蛋白,使用BCA试剂盒测定蛋白浓度。30 μg蛋白上样,经SDS-PAGE电泳分离后,采用湿转法将蛋白转移至PVDF膜上。5%脱脂奶粉封闭1 h,一抗4℃孵育过夜,二抗孵育2 h。使用ECL发光检测试剂盒进行显色,曝光。使用Image J软件对蛋白灰度进行半定量分析。
1.2.4 构建过表达CCKAR细胞株 使用SuperScriptTMⅣ第一链合成系统获得CCKAR cDNA,按照说明书将cDNA插入到pcDNA3.1质粒载体上,形成pcDNA3.1-CCKAR过表达载体,再依据Lipofectamine 2000说明书将过表达载体转染至BEAS-2B细胞中。
1.2.5 ELISA检测炎症因子水平 依据说明书使用ELISA试剂盒检测IL-1β、IL-6、TNF-α在细胞上清培养液中的分泌水平。
1.2.6 生物信息学分析 通过GEO数据库(https://www.ncbi.nlm.nih.gov/geo/)获取GSE18341和GSE2411表达数据。选取GSE18341芯片中未处理组小鼠(4个样本;16周)和LPS诱导组小鼠(4个样本),以及GSE2411芯片中Control组小鼠作为对照组(6个样本)和LPS组小鼠作为实验组(6个样本),应用GEO在线分析工具GEO2R对各自芯片中的对照组和试验组进行差异分析,以|logFC| > 1.5,P<0.05为筛选条件获得差异表达基因(differentially expressed genes,DEGs)。
1.3 统计学分析

2 结果
2.1 生物信息学预测结果
通过GEO数据库平台检测,GSE18341和GSE2411数据集符合研究标准。对两个数据集进行GEO2R差异分析,分别筛选出238个和120个DEGs,其中GSE18341有220个上调基因,18个下调基因(图1A,P<0.05);GSE2411有116个上调基因,4个下调基因(图1B,P<0.05)。利用韦恩图对两个数据集获得的DEGs取交集,发现有80个共同DEGs(图1C)。CCKAR是共同下调表达基因,并且在LPS诱导的小鼠急性肺损伤中下调最为显著。

A:GSE18341中DEGs火山图;B:GSE2411中DEGs火山图;C:GSE18341和GSE2411 DEGs的韦恩图图1 GSE18341和GSE2411中差异表达基因Fig.1 Identification of differentially expressed genes(DEGs)in GSE18341 and GSE2411
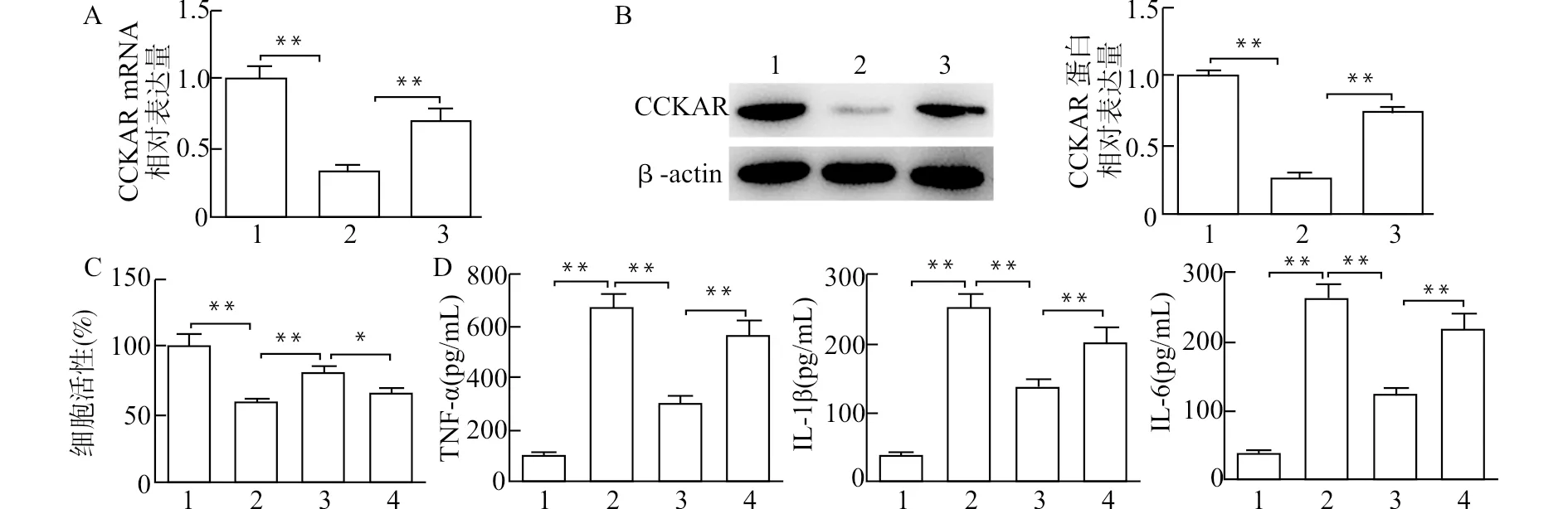
2.2 过表达CCKAR降低LPS诱导的BEAS-2B细胞存活率和炎症反应
RT-PCR和Western blot结果显示,BEAS-2B细胞经LPS诱导24 h后,CCKAR表达水平下调(图2A、2B,P<0.01),并且细胞存活率显著下降(图2C,P<0.01);与LPS+OE-NC组相比,CCKAR在LPS+OE-CCKAR组中的表达水平显著升高(图2D、2E,均P<0.01),并且细胞存活率显著上调(图2C,P<0.05)。ELISA结果表明,LPS诱导后,BEAS-2B细胞中炎症因子IL-1β、IL-6、TNF-α分泌表达水平显著上调(图2F,均P<0.01);与LPS+OE-NC组相比,CCKAR过表达后LPS诱导的炎症因子表达水平明显下调(图2F,均P<0.01)。

1:空白对照组;2:LPS组;3:LPS+OE-NC组;4:LPS+OE-CCKAR组;A:RT-PCR检测CCKAR mRNA表达水平;B:Western blot检测CCKAR蛋白表达水平;C:MTT检测细胞活力;D:RT-PCR检测CCKAR mRNA表达水平;E:Western blot检测CCKAR蛋白表达水平;F:ELISA检测TNF-α、IL-1β和IL-6和水平;*P<0.05 **P<0.01图2 CCKAR在LPS诱导的细胞损伤及炎症反应中的作用Fig.2 Role of CCKAR in LPS-induced cell injury and inflammation
2.3 过表达CCKAR激活NF-κB信号通路
与空白对照组相比,LPS诱导后p-p65/p65和p-IκB/IκB表达比值显著上调(图3,均P<0.01),而过表达CCKAR显著抑制LPS诱导的BEAS-2B细胞中的p-p65/p65和p-IκB/IκB表达比值(图3,均P<0.01)。
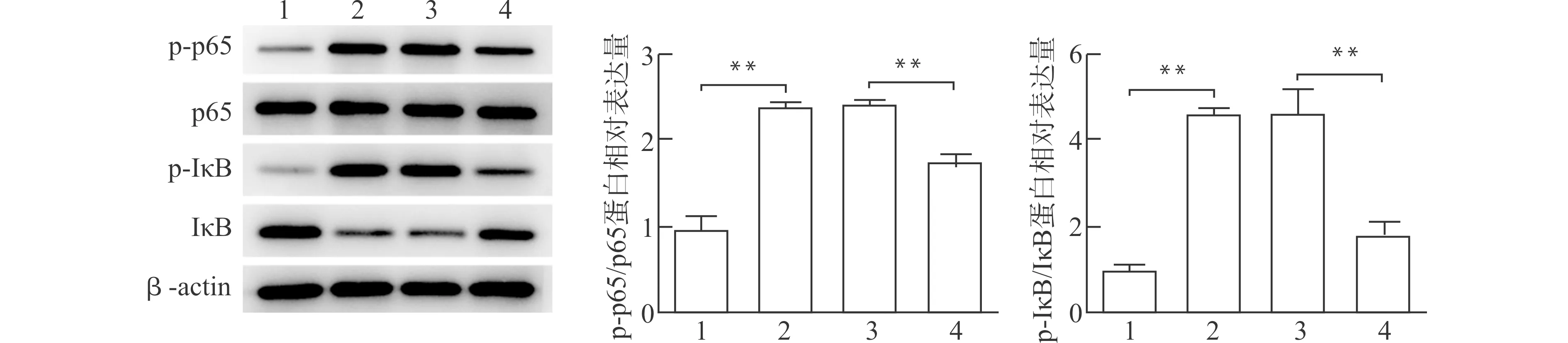
1:空白对照组;2:LPS组;3:LPS+OE-NC组;4:LPS+OE-CCKAR组;**P<0.01图3 CCKAR在LPS诱导的细胞对NF-κB信号通路的影响Fig.3 Effect of CCKAR on NF-κB signaling pathway in LPS-induced cells
2.4 CCK通过上调CCKAR减弱LPS诱导的细胞损伤及炎症反应
RT-PCR和Western blot结果显示,与LPS单独诱导组相比,LPS和CCK共处理导致CCKAR表达水平显著上升(图4A、4B,均P<0.01)。同时,CCK处理显著提高细胞存活率,抑制LPS对细胞存活率的影响(图4C,P<0.01);而CCK和CCK拮抗剂Proglumide共处理导致细胞存活率下降(图4C,P<0.05)。CCK处理后有效抑制了LPS引起的炎症因子IL-1β、IL-6、TNF-α表达上调(图4D,均P<0.01);而同时加入Proglumide后,CCK对炎症因子水平的影响受到抑制(图4D,均P<0.01)。
1:空白对照组;2:LPS组;3:LPS+OE-CCK组;4:LPS+CCK+Proglumide组;A:RT-PCR检测CCKAR mRNA表达水平;B:Western blot检测CCKAR蛋白表达水平;C:MTT检测细胞活力;D:ELISA检测TNF-α、IL-1β、IL-6水平;*P<0.05 **P<0.01图4 CCK通过CCKAR减弱LPS诱导的细胞损伤和炎症反应Fig.4 CCK reduces LPS-induced cell injury and inflammation through CCKAR
2.5 CCK通过上调CCKAR激活NF-κB信号通路
Western blot结果显示(图5),与空白对照组比较,加入LPS后,p-p65/p65和p-IκB/IκB表达比值均显著升高(均P<0.01);与LPS处理组比较,LPS+OE-CCK组p-p65/p65和p-IκB/IκB表达比值显著降低(均P<0.01),而在加入CCK拮抗剂后,p-p65/p65和p-IκB/IκB表达比值显著上调(均P<0.01)。

1:空白对照组;2:LPS组;3:LPS+OE-CCK组;4:LPS+CCK+Proglumide组;**P<0.01图5 CCK对CCKAR激活NF-κB信号通路的影响Fig.5 Effect of CCK on activation of NF-κB signaling pathway by CCKAR
3 讨论
ALI指在严重感染、休克创伤以及烧伤等非心源性疾病过程中,肺泡上皮细胞及肺毛细血管内皮细胞受到损伤,从而引起弥漫性肺间质和肺泡水肿,导致呼吸衰竭。ALI发展进程中,过度炎症级联反应会导致肺泡-毛细血管结构的完整性破坏。新生儿ARDS病理生理过程中也存在炎症现象[10]。肺上皮细胞是炎症损伤中重要的靶细胞,内毒素(LPS)是组成革兰阴性细菌细胞壁的成分,是刺激ALI等严重炎性疾病的主要因素。因而本文利用LPS诱导肺上皮体外模拟ALI,也可以一定程度上模拟新生儿ARDS。
GEO数据库收录了世界各国研究人员提供的高通量数据,是生物信息学分析研究的重要数据提取来源,为寻求疾病发病机制以及靶点筛选提供了重要依据[11]。此次研究通过数据库筛选出80个ALI样本与正常样本之间的差异表达基因,其中CCKAR为CCK靶点,在炎症过程中同样发挥重要作用,且有研究发现八肽缩胆囊素(cholecystokinin-8,CCK-8)能够减轻LPS诱导大鼠ALI过程中的炎症反应[12]。故而将CCKAR作为此次研究的切入点。
与健康人群相比较,ARDS患者中促炎因子IL-1β、IL-6、TNF-α的表达水平较高[13],而炎症反应可作为ALI发展过程中的驱动因素[14]。此次研究发现,经LPS诱导后,BEAS-2B细胞中炎症表达因子上调,且CCKAR表达下调。CCKAR是一个G蛋白偶联受体,可通过胞外信号激活发挥抗炎作用[15]。前期研究表明,抑制CCKAR能促进炎症因子如TNF-α、IL-6的产生[16-17]。而本次实验结果发现,过表达CCKAR可减轻LPS诱导的炎症反应。CCK家族参与抗炎和抗内毒素休克过程[18],在大鼠ALI研究中发现其能够减轻LPS诱导的ALI[12]。本实验发现,CCK处理后减轻LPS诱导的炎症反应,且上调CCKAR表达;而CCK拮抗剂处理下调CCKAR表达并促进炎症反应,表明CCK可能通过刺激CCKAR表达而降低炎症反应,这与之前的研究结果一致[8-19]。此外,CCK处理后,细胞中NF-κB信号通路被抑制,而在同时加入CCK拮抗剂后,NF-κB信号恢复活性。NF-κB是参与炎症反应的重要信号通路,有研究发现,NF-κB影响ALI中的炎症、细胞凋亡以及氧化应激反应[20-21]。NF-κB信号可调节肺内促炎因子的释放,在各种致病因子如LPS作用下,NF-κB的过度活化促进炎症进程,在ALI/ARS中发挥重要作用[22-23]。IκB是NF-κB二聚体的抑制蛋白家族,IκB通过与NF-κB结合阻止其向核内转移发生活化,而磷酸化IκB可以解离NF-κB,进而促进其活化,进一步启动炎症基因转录[24]。研究发现,在LPS诱导的ALI模型中,抑制IκB可以抑制NF-κB的活化进而缓解急性肺损伤[25]。在血管上皮细胞中,CCK-8通过CCKAR和CCKBR显著抑制LPS诱导的NF-κB的活化[26]。
本次研究显示,上调CCKAR能够抑制NF-κB信号通路的激活,进而影响炎症反应,而CCK能够上调CCKAR的表达。而在一些研究中也发现,CCKAR激活能够抑制LPS诱导的NF-κB信号通路的激活[27]。由此表明,CCK可能通过CCKAR抑制NF-κB信号通路的激活进而影响炎症反应的发生。
新生儿ARDS严重威胁新生儿生命,多见于足月儿重症肺炎或败血症等重症感染[28-29]。ARDS同样可发生在早产儿身上,且不同于我们已经熟知的常见于早产儿的新生儿呼吸窘迫综合征。严重肺部感染以及脓毒症等可直接或间接引发新生儿ARDS,是威胁新生儿生命的危重疾病[30]。有研究发现在新生小猪ARDS模型中,抑制肺泡上皮细胞过度炎症对保护新生小猪肺部有重要意义[31]。本课题组研究发现CCKAR在LPS诱导的炎症反应过程中发挥作用,而抑制CCKAR可以抑制炎症反应。因而我们推测其在新生儿ARDS中也发挥一定作用,为后期在新生儿ALI/ARDS中的研究以及以该受体为靶点开发药物提供了相关依据。
