Natural history and management or liver aySuncuon ystorage disorders
2022-11-29MoinakSenSarmaParijatRamTripathi
Moinak Sen Sarma,Parijat Ram Tripathi
Abstract
Lysosomal storage disorders (LSD) are a rare group of genetic disorders. The major LSDs that cause liver dysfunction are disorders of sphingolipid lipid storage [Gaucher disease (GD) and Niemann-Pick disease] and lysosomal acid lipase deficiency [cholesteryl ester storage disease and Wolman disease (WD)]. These diseases can cause significant liver problems ranging from asymptomatic hepatomegaly to cirrhosis and portal hypertension. Abnormal storage cells initiate hepatic fibrosis in sphingolipid disorders. Dyslipidemia causes micronodular cirrhosis in lipid storage disorders. These disorders must be keenly differentiated from other chronic liver diseases and non-alcoholic steatohepatitis that affect children and young adults. GD, Niemann-Pick type C, and WD also cause neonatal cholestasis and infantile liver failure. Genotype and liver phenotype correlation is variable in these conditions. Patients with LSD may survive up to 4-5 decades except for those with neonatal onset disease. The diagnosis of all LSD is based on enzymatic activity, tissue histology, and genetic testing. Enzyme replacement is possible in GD and Niemann-Pick types A and B though there are major limitations in the outcome. Those that progress invariably require liver transplantation with variable outcomes. The prognosis of Niemann-Pick type C and WD is universally poor. Enzyme replacement therapy has a promising role in cholesteryl ester storage disease. This review attempts to outline the natural history of these disorders from a hepatologist’s perspective to increase awareness and facilitate better management of these rare disorders.
Key Words: Lysosomal; Gaucher; Niemann-Pick; Wolman; Cholesteryl ester; Children
INTRODUCTION
Lysosomes are intracellular organelles that contain multiple enzymes required for the degradation of a range of macromolecules. These enzymes have acidic pH and hydrolyze mucopolysaccharides, glycosphingolipids, and oligosaccharides. Each enzyme is specific for a particular molecule and essential for its catabolism. Lysosomal storage disorders (LSDs) arise from the defect in these enzymes, culminating into the specific substrate accumulation in the lysosomes, which finally causes cellular dysfunction.
Patients with LSDs have variable ages of onset ranging from the perinatal period to adulthood. These are a heterogeneous group of genetic disorders with multisystem involvement. Symptoms vary depending on the most affected organ systems. The most common symptoms are coarse facial features, skeletal dysplasia, hepatosplenomegaly with liver dysfunction, and neuroregression. The combined prevalence of LSDs is 1 per 7000 live births although individual disorders are uncommon[1,2].
There are more than 50 LSDs identified[1]. The classification and cardinal features of some of the important disorders are shown in Table 1. Mucopolysaccharidoses, mucolipidoses, and glycoprotein storage disorders only cause hepatomegaly without causing liver dysfunction. Disorders of sphingolipid and lipid storage disorders such as Gaucher disease (GD), Niemann-Pick disease (NPD), and lysosomal acid lipase deficiency (LAL-D) cause liver diseases ranging from asymptomatic hepatomegaly to cirrhosis and portal hypertension. All these disorders present a different set of challenges as they mimic other liver diseases. Their diagnosis requires enzyme analysis and genetic tests. Therapy is limited and response is variable if treatment exists. Simultaneous involvement of other organ systems often precludes the possibility of liver transplantation (LT). Our review aims to describe the natural history of the liver disease in LSDs. The review is focused on the LSDs that have significant liver dysfunction such as GD, NPD, and LAL-D. The novelty of the review is to comprehensively collate the available literature so that hepatologists will have a better understanding of the disease management.
GD
General aspects
GD is a disorder of sphingolipid storage occurring as a result of the deficiency of acid β-glucosidase enzyme in the nucleated cells. There is defective cleavage of glucosylceramide and glucosylsphingosine, resulting in their accumulation in lysosomes. There is infiltration of macrophages laden with glucosylceramide in visceral tissues such as the bone marrow, spleen, liver, and lymph nodes (Figure 1). These cells, “foamy macrophages” or “Gaucher cells (GC)”, have a signet ring with a crumpled-paper appearance as the nucleus is pushed to one side (Figure 2). Neurological involvement is mainly due to damage by the lipids directly rather than the infiltration by foamy macrophages. GD is more common than other LSDs, with a high prevalence of approximately 1 in 855 individuals in the Ashkenazi Jewish population[3].
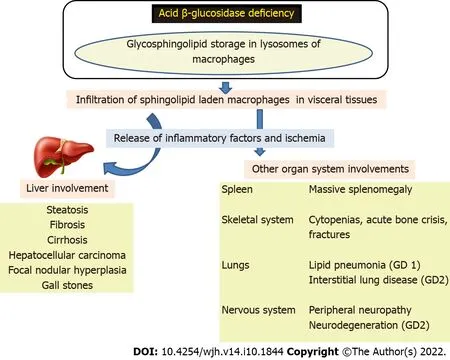
Figure 1 Pathogenesis of Gaucher disease. GD: Gaucher disease.
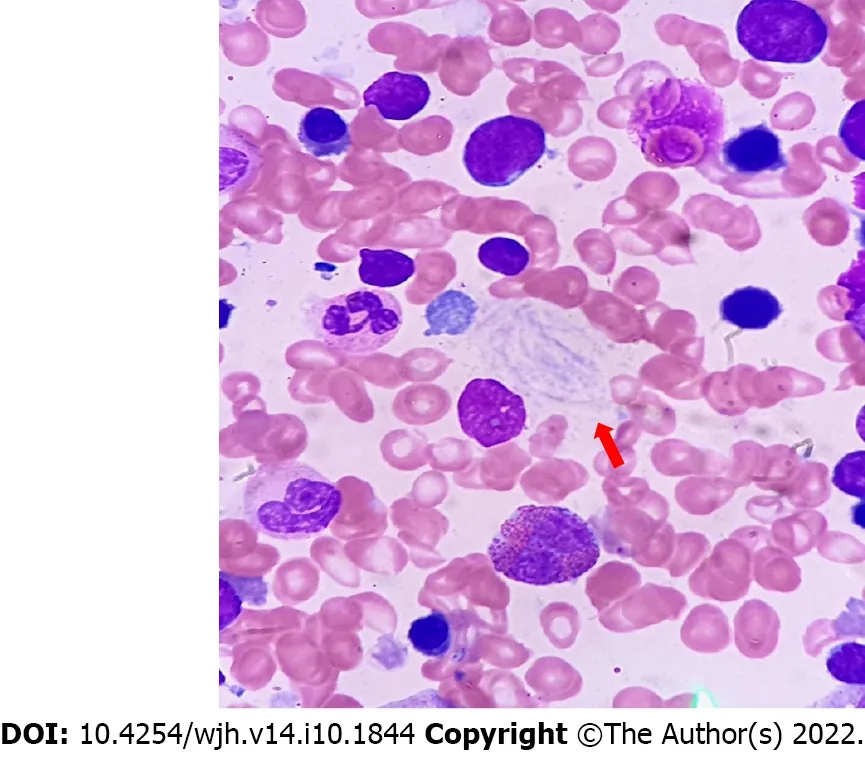
Figure 2 Histology of Gaucher disease. Red arrow shows Gaucher cells.
Depending on neurological involvement, GD is divided into three types: Type 1, no neurological involvement, with a prevalence of 1/40000; Type 2, neurological involvement in infancy, with a prevalence of < 1/100000; Type 3, variable neurological manifestations, with a prevalence of < 1/50000 to < 1/100000[4].
Clinical manifestations and natural history of GD
There are two phenotypes of liver presentation. The first one is milder with hepatomegaly, nonmalignant focal liver lesions, and fibrosis. The other is severe, presenting as cirrhosis, portal hypertension, and potential hepatocellular carcinoma (HCC).
Hepatomegaly occurs as a result of the infiltration of GC in the liver and macrophage lipid accumulation. Liver volumes > 1.25 and 2.5 or > 2.5 multiples of normal are classified as mild, moderate, and severe hepatomegaly, respectively. Moderate or severe hepatomegaly at diagnosis is seen in approximately 80% of patients. Mean liver and spleen volumes are 1.8 and 19.4 times of normal in untreated GD subjects[5]. Hepatomegaly is less massive than splenomegaly in GD[6]. If the liver size outweighs the splenic size, one must carefully evaluate the other causes of liver disease or concurrent comorbidities[7]. Various modalities of assessment report the sizes of the organs variably. Sonographicprevalence of hepatomegaly was noted in 100% of pediatric patients[8]. Magnetic resonance imaging (MRI) has shown that 77%-95% of adults suffering from GD have hepatomegaly of variable degrees[9]. Biochemical liver dysfunction is noted in 19%-55%. Hepatic involvement in GD may lead to portal hypertension and endstage liver disease. Massive splenomegaly can also produce increased portalflow leading to pre-hepatic portal hypertension and overestimation of the degree of liver disease[10]. The decision for splenectomy as a therapeutic intervention is delicate given the other comorbidities that ensue in splenectomized patients over time[11].
During imaging, multiple hypoechoic and/or hyperechoic lesions are seen in the liver and spleen. Focal hyperechoic liver lesions can be seen in 5% of individuals on sonolography[12]. Small lesions reflect a focal accumulation of GC[13]. These lesions do not merit biopsy as the growth will be slow. Approximately 20% have early focal signal abnormalities on MRI (hypointense on T1, and heterogeneous on T2) and are hypoattenuating on computed tomography in GD[9]. These lesions do not respond to enzyme replacement therapy (ERT) but they must be kept on surveillance. Enlarging lesions on follow-up with rising serum alpha-fetoprotein is a concern. Focal nodular hyperplasia has been reported in GD[14]. HCC is more common in the setting of advanced liver disease with cirrhosis, in the pre-ERT era and splenectomized patients, and also in those with concomitant iron overload[15,16].
Due to bile lipid composition abnormalities, cholesterol gall stones have been reported in GD patients[17]. The reasons for increased stone predilection in splenectomised patients are unclear. Autoimmune hepatitis in the setting of GD has a guarded outcome and requires LT[18]. Viral hepatitis resulting from transfusion dependency, surgical interventions, and intravenous ERT is a serious concern. Chronic viral hepatitis B and C should be screened at baseline and at follow-up visits in patients with GD[19]. During therapy of concurrent chronic viral hepatitis, GD-related cytopenias may be confounders to anti-viral therapy. Initiating ERT to reduce the bulk of the disease, normalization of counts followed by antiviral therapy is a safe and rational approach[20]. In those with transfusion-related hepatitis C, the present newer antiviral drugs are not associated with cytopenias.
Transient elastography though recommended has no age-specific nomograms for interpretation. It may not distinguish steatosis from infiltrative fibrosis. Liver stiffness is mildly elevated in GD without cirrhosis and those with ERT-related reductions in liver fibrosis[1]. Those with cirrhosis exhibit significant liver stiffness. Since splenectomized patients have a higher degree of disease, their liver stiffness is greater as compared to those with mild disease[2]. Gaucher clinical severity (GD-DS3) scores also correlate well with MRI-measured liver stiffness[21]. Hence, this suggests that elastography may have a significant clinical utility in the management of GD. Abdominal paracentesis is best avoided from the left side due to massive splenomegaly or cautiously performed under sonolographical guidance. The hepatic venous pressure gradient is important to distinguish pre-portal hypertension from cirrhosis. Due to severe hypersplenism, transjugular liver biopsy is often warranted over percutaneous biopsy.
Neonatal cholestasis is an important presentation in GD. Soudek et al[22] reported a 4-day-old neonate with GD who presented with cholestasis, low glucocerebrosidase activity (2 nmol/h/mg protein), and deletion of exons 3-12 and c. 1448T>C (p.Leu483Pro) in the GBA gene. After failed ERT, the liver dysfunction progressed. LT was performed at 7 mo and liver functions improved. The child died later due to progressive neurological involvement. A review of the literature was performed with nine other previously reported cases. The median age of presentation was 1 (1-51) d of life. The age at diagnosis was 3 (1-6.5) mo. The mean value for total bilirubin was 284 [interquartile range (IQR): 166-304] μmol/L, direct bilirubin was 117 (IQR: 92-265) μmol/L, aspartate transaminase (AST) was 514 (IQR: 420-670) IU/L, alanine transaminase (ALT) was 280 (IQR: 206-441) IU/L, and gamma-glutamyl transpeptidase (GGT) was 208 (IQR: 165-242) IU/L. Six of these patients had neurological presentations by the ages of 2 (0.4-10) mo. Bone marrow and liver biopsies had poor yield. Some of the patients even required a second biopsy for confirmation. The common genetic mutations were c.1342 G>C (p.Asp448His) and c. 1448T>C (p.Leu483Pro). Of the four patients who received ERT, two showed an improvement in bilirubin and platelets. Over a limited follow-up period of 6 mo, the rest seven patients died by the age of 4.75 (IQR: 4-13) mo. Five died due to respiratory causes and two due to gastrointestinal bleeding. Long-term outcomes in this phenotype are not known[23]. There may be an ethical dilemma of LT in children with decompensated liver disease before a fully evolved neurological disease. Parental consent is required in such scenarios.
Liver pathology in GD
GC is characterized by lightly striated eosinophilic cytoplasm and small round vesicular nuclei. There may be some accumulation of intracellular iron. GC has predilection for central zonal distribution (zone 1). Hepatocytes do not accumulate glycosphingolipids as such. However, in close proximity to a cluster of GC, it may undergo degenerative changes. In GD, liver histology shows a wide range of features. In milder disease, there are scattered foci of GC with mild structural parenchymal changes. In advanced cases, there is cirrhosis with dense infiltration of the liver by GC. Most cases have pericellularfibrosis, and 20%-50% have bridging or more severefibrosis[23]. GC induces inflammatory factors that promote thefibrogenic process. Gaucheromas are large clumps of GC with areas offibrosis[24-26]. In a single needle biopsy, Gaucheromas pose a diagnostic challenge since they can mimic liver malignancies and have considerable radiological dilemmas[27,28]. Diffuse steatosis in GD occurs as a part of a metabolic syndrome which occurs either de novo or as a side effect of long-term ERT. In GD, dyslipidemia and biliary lipid secretion abnormalities can occur[29,30]. Mouse models suggest that suppression of GC levels may be associated with a rise in glycolipids and metabolic derangements[31]. Hepaticfibrosis occurs due to GC infiltration and diffuse low-grade inflammatory processes caused by the activated macrophages or GC[32]. Hepatic microinfarcts result from larger clusters of GC which promote liverfibrosis[33]. Splenectomy may cause liver injury due to the shift of balance of the spleen as the preferred storage organ. Secondary hemochromatosis has been reported due to iron deposits in hepatocytes and Kupffer cells in GD. Elevated ferritin levels reflect iron overload and chronic inflammation[34,35]. Studies in Ashkenazi Jews and animal models have revealed that higher levels of GC may be hepatoprotective in those with liver comorbidities such as hepatitis B and C and non-alcoholic steatohepatitis. Several mechanisms have been postulated to explain the hepatoprotective nature of GC. GC may serve as a glycolipid ligand and is presented to non-killer T cells and dendritic cells via CD1 molecules[36]. By changing the cross-talk between these cells and other immune system cells, GC can exert an immunomodulatory effect directly or indirectly on these target cells. They also alter lipid rafts and intracellular signaling machinery, promote regulatory T lymphocytes, and improve immunogenicity. They may function as metabolic intermediates in insulin resistance and promote mucosal immunity[37]. Alpha-glycolipids are hepatotoxic but β-glycolipids are hepatoprotective[38]. Liver biopsy showing GC should be distinguished from liver “pseudo-GC” which is better appreciated in an additional bone marrow examination. The pseudo-GC has been demonstrated in acute lymphoblastic leukemia, myelodysplasia, Hodgkin's disease, thalassemia, and disseminated tuberculosis[39].
Therapy and LT in GD
Therapeutic approaches are ERT or substrate reduction therapy (SRT). Those who are on specific ERT or SRT will most likely experience reductions in liver dysfunction and sizes of organomegaly within 6-12 mo. The newly FDA-approved eliglustat, as a first-line option for GD, can improve liver fibrosis. Conversely, hepatic fibrosis may progress despite high-dose ERT. Advanced liver disease invariably requires LT. Ayto et al[18] reviewed outcomes in patients with GD undergoing LT. Good outcomes of LT with concurrent ERT were reported. There was no evidence of GD-related pathology in the liver graft even at 10 years of follow-up. In very rare cases, splenectomy can be considered for portal hypertension if cirrhosis is ruled out.
NPD TYPES A AND B
General aspects
NPD has three types: A, B, and C. The prevalence of NPD-A and NPD-B is 1 in 250 000, which is even higher in Ashkenazi Jews, where it is 1 in 40 000[40]. NPD-A and NPD-B are caused by acid sphingomyelinase deficiency (ASMD). Acid sphingomyelinase cleaves sphingomyelin into ceramide and phosphocholine and its deficiency leads to excessive accumulation of sphingomyelin and its precursor lipids (Figure 3). Foamy histiocytes, which are the characteristic storage cells of NPD, accumulate in visceral organs like the liver, spleen, bone marrow, lungs, and kidneys. Cherry red spots may be seen in the eyes (Figure 4). In NPD-A patients, sphingomyelin is also accumulated in the brain. NPD-A and NPD-B represent different phenotypic spectrums of the same disease. Genetic testing is the gold standard for confirmation of a diagnosis of all NPD. From a hepatologist’s point of view, NPD-B and NPD-C are most important for management. NPD-C will be discussed separately due to its unique presentation.
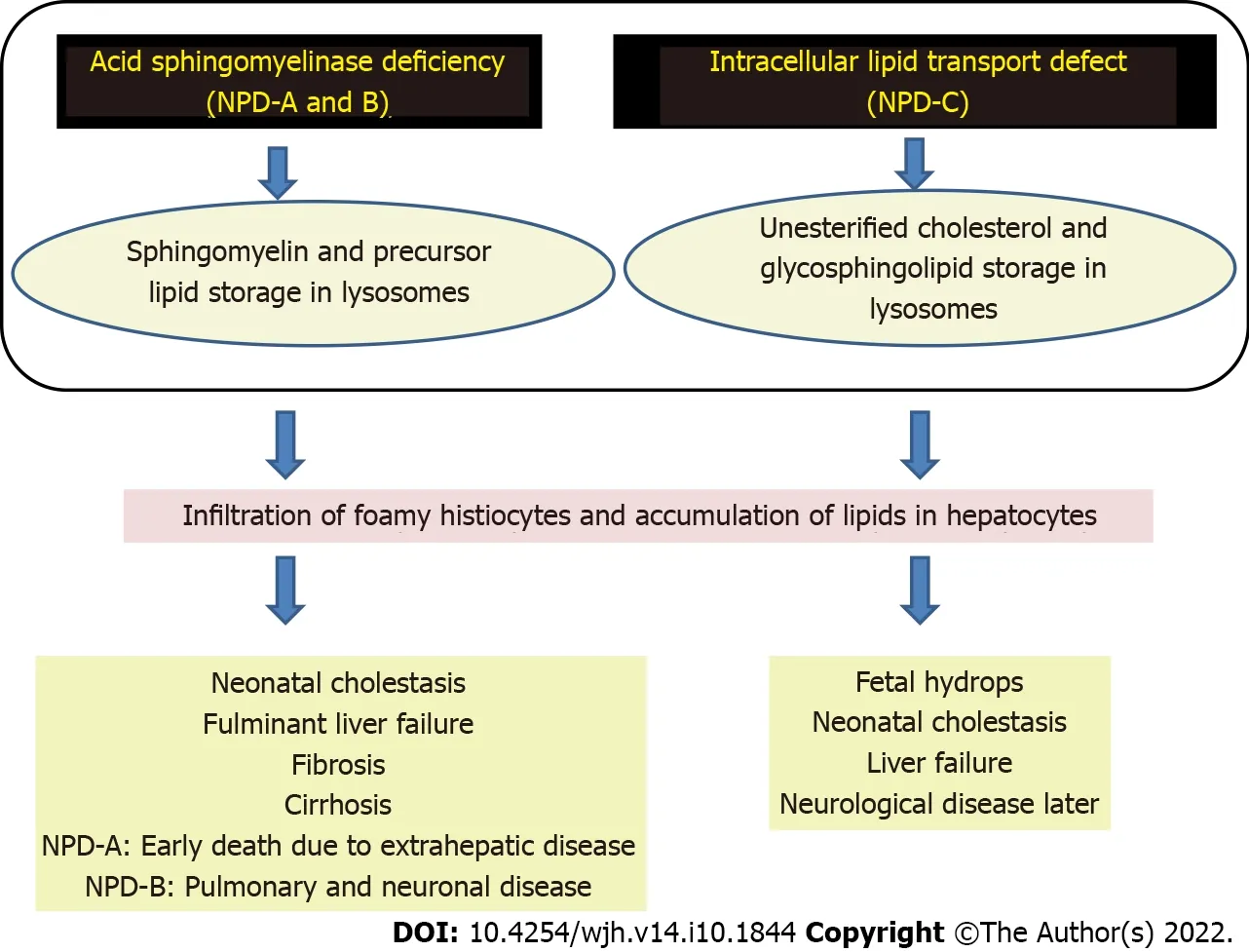
Figure 3 Pathogenesis of Niemann-Pick disease types A, B, and C. NPD: Niemann-Pick disease.
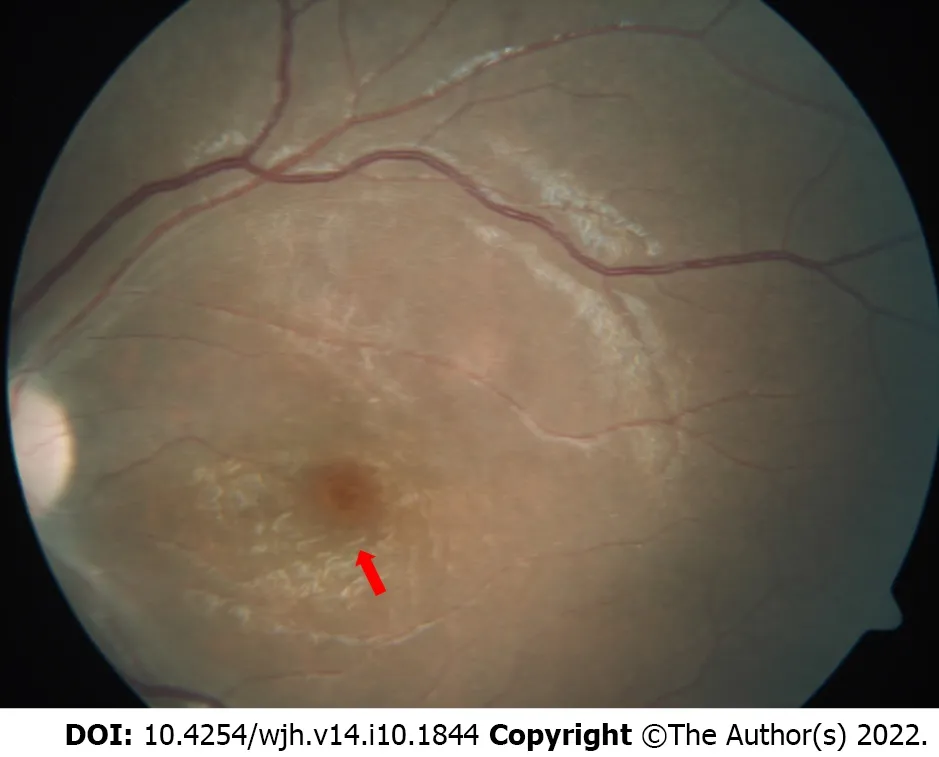
Figure 4 Fundus examination showing a cherry red spot (red arrow) with a background of white retina.
Clinical manifestations and natural history of ASMD
As is well known, NPD-A presents as infantile onset and such patients die early in childhood without much progress in the liver disease. Deaths in NPD-A patients occur predominantly due to extrahepatic manifestations. Terminal liver disease occurs very rarely in NPD-A. NPD-B presents as infantile to adult onset with a significant proportion having progressive liver dysfunction and approximately 40% requiring LT. Those who survive with their native liver die due to pulmonary or neurological disease[40].
McGovern et al[41] showed that baseline mean liver volume was 2.1 ± 0.8 times that of normal (MN) (range 0.9-4.6) in ASMD patients. In children, the mean liver volume was 2.2 ± 0.7 MN at baseline, 2.1 ± 0.7 at 1 year, and 1.7 ± 0.4 at the final visit, showing that there was minimal appreciable change. In adults, liver volume is 1.9 ± 0.9 MN at baseline, 1.6 ± 0.5 at 1 year, and 1.5 ± 0.4 at the final visit, showing some reductions over time. However, moderate to severe hepatomegaly (1.25-1.75 MN) was observed in 96% at baseline, 97% at 1 year, and 88% at the final visit. In NPD-B, hepatomegaly (mean liver volume is 1.9 MN) affects up to 70% of patients, less severe than splenomegaly (mean spleen volume is 11.1 MN). Liver volume correlates with splenic volume and severity of extra-hepatic manifestations of NPD-B[42]. In a prospective multicenter longitudinal study, it was shown that ALT and AST were high in 47% and 51% of individuals, respectively. Mean baseline ALT was 86.2 ± 67.8 U/L and 51.5 ± 44.6 U/L in children and adults, respectively. Mean ALT values at the final follow-up were 65.5 U/L in children and 52.7 U/L in adults. Total bilirubin was elevated in 33% at baseline, similar in adults and children (17.5 vs 21.8 mg/dL, respectively) and similar at follow-up. Synthetic functions (prothrombin time, platelet count, and albumin level) remained stable or worsened during the study[43-45].Wasserstein et al[43] followed 29 patients with NPD-B for 10 years and analyzed liver function at baseline and maximum follow-up visits (at least 9 mo or more apart). In the natural history of these patients (2-64 years of age at study entry), liver dysfunction was common. Approximately 75% of patients had elevated ALT and 65% had elevated AST at the initial visit. At baseline, there were no significant differences in liver function with respect to gender or age. However, total bilirubin was higher in patients > 18 years old. Similarly, there were no statistically significant changes in liver function during the follow-up period. Liver enzymes remained high whereas bilirubin remained normal in most patients throughout the study.
In a single center study of 103 patients with NPD-B over 10 years, six patients had fulminant liver failure, and three showed evidence of cirrhosis on liver biopsy. Two of the patients with liver failure received successful orthotopic LT at 12 and 25 years of age while the rest died from liver failure[42]. In another study by Wasserstein et al[43], one patient developed hepatic dysfunction in the first decade and subsequently died of liver failure. Homozygotes for R608, P323A, and P330R had milder disease than other genotypes. In another case report, an adult died at the age of 31 years from refractory encephalopathy related to cirrhosis and hepatic failure[44]. In 13 Chilean children homozygous for the SMPD1 p. (Ala359Asp) (A359D) mutation (associated with moderate to severe NPD B), five patients developed progressive cirrhosis. All five patients had sustained approximately four-fold increases in liver enzymes. Three of these patients died of liver failure and the other two received LT[45]. Cassiman et al[46] collected the data from 85 patients who died from NPD type B. They had splenomegaly (96.6%), hepatomegaly (91.4%), liver dysfunction (82.6%), and pulmonary involvement (75.0%). The median age atfirst symptom onset, age at diagnosis, and age at death or LT were 0.8 (0-60), 2.0 (0.2-78), and 18 (0.58-78) years, respectively. The leading causes of death were respiratory and liver failure (27.7% each) irrespective of age. The authors divided their cohort as chronic visceral vs neurovisceral ASMD. In the analysis, chronic visceral ASMD had lower age atfirst symptom onset (0.5 vs 1.25 years), diagnosis (1.7 vs 5 years), and death or LT (8 vs 23.5 years). Compared to chronic neurovisceral ASMD, 31.8% had progression of neurodegenerative disease along with respiratory disease (both 23.1%) and liver disease (19.2%) leading to death. In the subgroup of 23 patients with terminal liver disease, age of symptoms onset was 0.8 (0.17-5) years and age at diagnosis was 3 (0.2-67) years. Twelve (52.2%; age range 2.5-18 years) and 11 patients (47.8%; age range 21-67 years) died or had LT in childhood. The overall median age at death was 18 (2.5-67) years. Other liver-related deaths were variceal bleeding (n = 4) and hepatocellular carcinoma (n = 2)[46]. McGovern et al[41] concluded that individuals with either severe spleno-megaly or prior splenectomy were a significant risk factor of death than those with smaller or intact spleens (odds ratio = 10.29, 95%CI: 1.7, 62.7).
Liver pathology in ASMD
Sphingomyelin accumulation in Kupffer cells and hepatocytes caused hepatomegaly. Hypertransaminasemia does not correlate with the stage of hepaticfibrosis or severity of the liver disease[48]. Evolution of fibrosis is variable. Among 17 NPD-B patients, 88% hadfibrosis and 12% progressed to cirrhosis[48]. Sphingomyelin accumulation in sinusoidal Kupffer cells shows an enlarged and foamy appearance (Figure 5). In NPD-B patients with liverfibrosis, the stored sphingomyelin is seen as large collections of foamy Kupffer cells in portal areas as well as within hepatocytes[49].
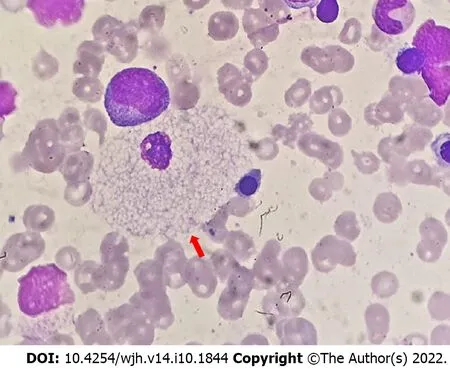
Figure 5 Histology in Niemann-Pick disease type B. Red arrow shows a foamy vacuolated histiocyte.
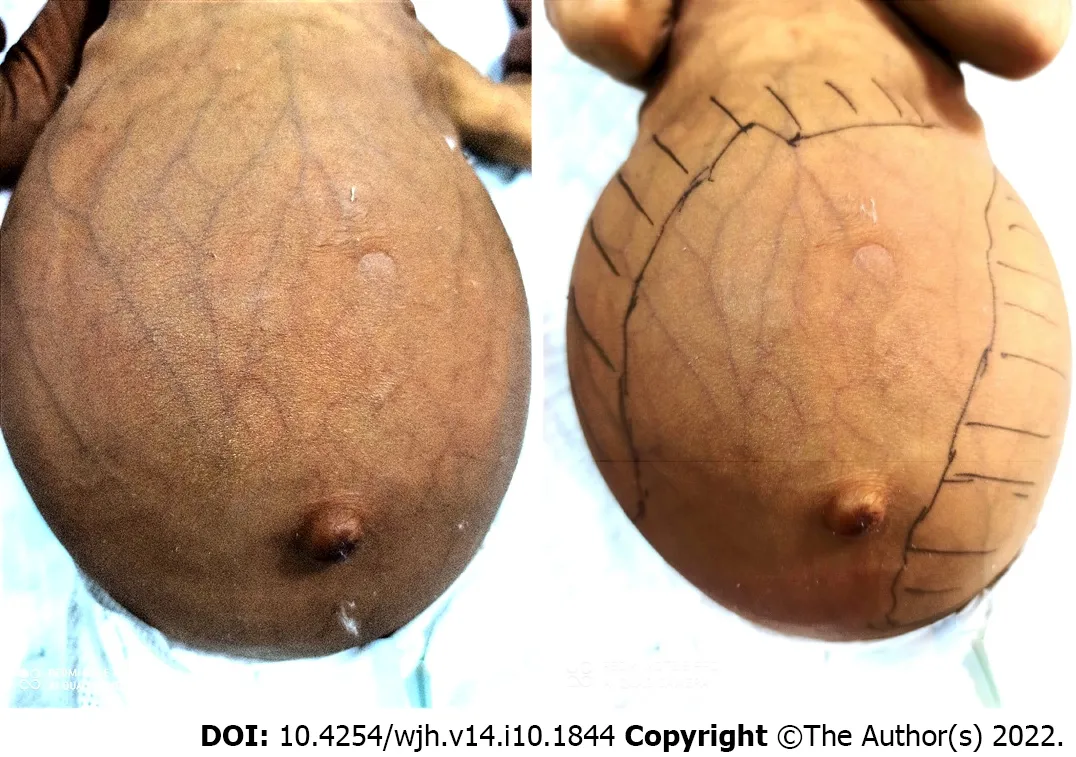
Figure 6 Infant with Niemann-Pick disease type C presenting as cholestasis, dilated abdominal veins, and massive hepatosplenomegaly.
Therapy and LT in ASMD
Currently, there is no disease-specific treatment for NPD-B. ERT with a recombinant human acid sphingomyelinase (olipudase alfa) is in clinical development. Olipudase alfa reduced liver and spleen volume by 31% and 39%, respectively, in a phase 2 trial evaluating five adults who were followed for 30 mo. It improved respiratory reserve by 35%, lipid profile, and bone health (bone mineral density in spine). Adverse events were headache, nausea, and abdominal pain. Anti-drug antibodies and hematological or cardiac side effects were not present. With olipudase alfa treatment, biomarkers such as chitotriosidase in serum and lysosphingomyelin in dried blood spots decreased remarkably[50]. Morbidity and disease burden are governed by respiratory disease and organomegaly in chronic ASMD. These are also independent contributors to mortality. The degree of splenomegaly correlates with short stature, atherogenic lipid profile, and hematological abnormalities. Respiratory-related complications are a major cause of mortality in ASMD. Hence Jones et al[51] concluded that lung function and spleen volume are meaningful clinical end points for assessing disease burden in ASMD.
Liu et al[52]performed LT in seven patients with NPD-B who were symptomatic at 12 (6-14) mo and transplanted at 6.5 (2.2-8.6) years. Among them, four patients received living donor LT, and three received whole-liver orthotopic LT. At a median follow-up of 10 (5-53) mo, all patients were alive with adequate catch-up growth. Liver function normalized within 3 wk after transplantation with improvement in platelet count, leukocyte count, and triglyceride levels. Pulmonary disease ameliorated after transplantation with resolution of interstitial lung disease and improved lung function. However, those with psychomotor improvement and developmental delays had persistent symptoms. The authors concluded that LT was an effective therapy for patients with NPD type B with severe liver and pulmonary dysfunction.
NPD TYPE C
NPD-C is categorized along with other NPDs due to the presence of foamy macrophages but there is no deficiency of acid sphingomyelinase enzyme. In NPD-C, there is an intracellular lipid trafficking defect. There is progressive lipid accumulation (unesterified cholesterol and glycosphingolipids) within the lysosomes. The prevalence of NPD-C is 1 in 120 000 live births. There is variable age of onset along with both visceral and neurological involvement. There are two types of NPD-C, type 1 (NPC1) and type 2 (NPC2). Both types have different genetic mutations, but clinical presentation is similar. NPC1 constitutes 95% while the remaining 5% are NPC2 patients. Elevated plasma chitotriosidase is a useful screening test in young children for NPD-C (and GD) but has a low sensitivity and specificity. Similarly, chemokine (C-C motif) ligand 18 (CCL18) is also a screening test. A positive Filipin stain of bone marrow is seen in 85% of NPD-C cases but it should not be considered a definitive assessment[41].
Age of presentation of NPD-C is variable from the perinatal period to the adult age. Broadly accepted age onset subgroups are perinatal (< 3 mo), early-infantile (3 mo to 24 mo), late-infantile (2 to 6 years), juvenile (6-15 years), and adolescent/adult (> 15 years). As a unique variant, NPD-C that starts early presents as neonatal cholestasis, infantile liver failure, ascites, or hydrops. This variant is very aggressive and the majority of patients have a poor prognosis[53]. In all types, the neurovisceral variant is more aggressive than visceral from the liver point of view. Splenectomy may worsen the liver, just like GD. Other than progressive liver disease, pulmonary complications are common. Most cases die of respiratory insufficiency or chest infections. Reasons for respiratory insufficiency are infiltration of the lungs with foamy cells, worsening organomegaly, and ascites. The respiratory complications are partly related to immune dysregulation in NPD-C[54]. Patients who survive beyond the first month of life without hepatic or respiratory failure, will eventually die of progressive neurological disease[55]. Castaneda et al[54] showed that none of the patients had neurological involvement at the time of diagnosis. The deceased patients with delayed developmental milestones have progressive deficits in ambulation, speech, swallowing, and feeding.
Neonatal liver disease has prominent hepatosplenomegaly in NPD-C (Figure 6). In one study, NPD-C accounted for 7.5% of all infants evaluated for cholestasis[56]. Ten patients with NPD-C had an age of onset and age at diagnosis of 3.6 (1-10) d and 14.6 (1-30) d, respectively. Total and conjugated bilirubin levels were 13.9 (8-23) mg/dL and 8 (3.4-13.5) mg/dL, respectively. The serum AST level was 300.2 (101-700) U/L, which was nearly three times the upper limit of normal[57]. In most cases, cholestasis is transient for a few months. Hepatosplenomegaly persists for a variable period before the onset of neurological symptoms. These manifestations are important clues toward a possible diagnosis of NPDC in infancy[58]. Isolated unexplained splenomegaly, with or without hepatomegaly, in a neonate or infant should raise suspicion of NPD-C[59]. Approximately 10% of cases progress to liver failure and usually die before the age of 6 mo in those with early prolonged jaundice and hepatosplenomegaly[54].
Prenatal onset NPD-C is a distinct and severe subgroup of the neonatal-onset NPD-C[60]. Fetal ascites or non-immune fetal hydrops can be seen in the perinatal period. In a study of seven NPD-C patients with prenatal manifestations, it was observed that these patients had a poor postnatal course. Of the two NPC1 patients who presented with fetal ascites at birth, one had prenatal ultrasonography at the 27thweek of gestation that showed hydrops fetalis and polyhydramnios. This child later died in the first year of life due to progressive liver failure and pulmonary insufficiency. The second patient with similar clinical findings survived without progressive ascites or liver failure. Siblings with the same molecular defect may have different disease outcomes and variable presentation and severity of perinatal onset NPD-C[61].
There is no curative therapy for NPD-C. Patients with NPD-C are largely managed with supportive treatment and multidisciplinary care. Miglustat can be used as a disease-modifying agent, which is approved by European Union. In a study on 29 patients, after 12 mo of therapy, there was an improvement in horizontal saccadic eye movements, swallowing reflex, auditory acuity, and ambulation[62]. Further extension of the same cohort with 2-year treatment has shown stabilization of neurological symptoms (cognition, ambulation, and swallowing)[63]. Miglustat is more effective in patients with late-onset neurological symptoms as compared to those with early-onset disease.
LAL-D
General aspects
Wolman disease (WD) and cholesteryl ester storage disease (CESD) represent the clinical spectrum of LAL-D. The incidences of WD and CESD are approximately 1:500 000 and 1:40 000, respectively[64]. There is a defect in the LIPA gene (chromosome 10q23) which encodes LAL. There is an almost complete deficiency of LAL in WD while in CESD, there is some residual activity. LAL-D leads to impaired metabolism of triglycerides and cholesteryl esters resulting in their accumulation in macrophages and lysosomes of hepatocytes (Figure 7)[65]. There are many clinical differences between CESD and WD. WD has onset by 3 mo age and usually dies within 6 mo, whereas CESD begins in childhood or adulthood and age at death is variable. Compared to mild or absent features in CESD, WD has marked hepatomegaly, splenomegaly, malabsorption, growth failure and dyslipidemia. And 50% of WD have adrenal calcification which is rarely found in CESD[66]. The two entities are described separately.
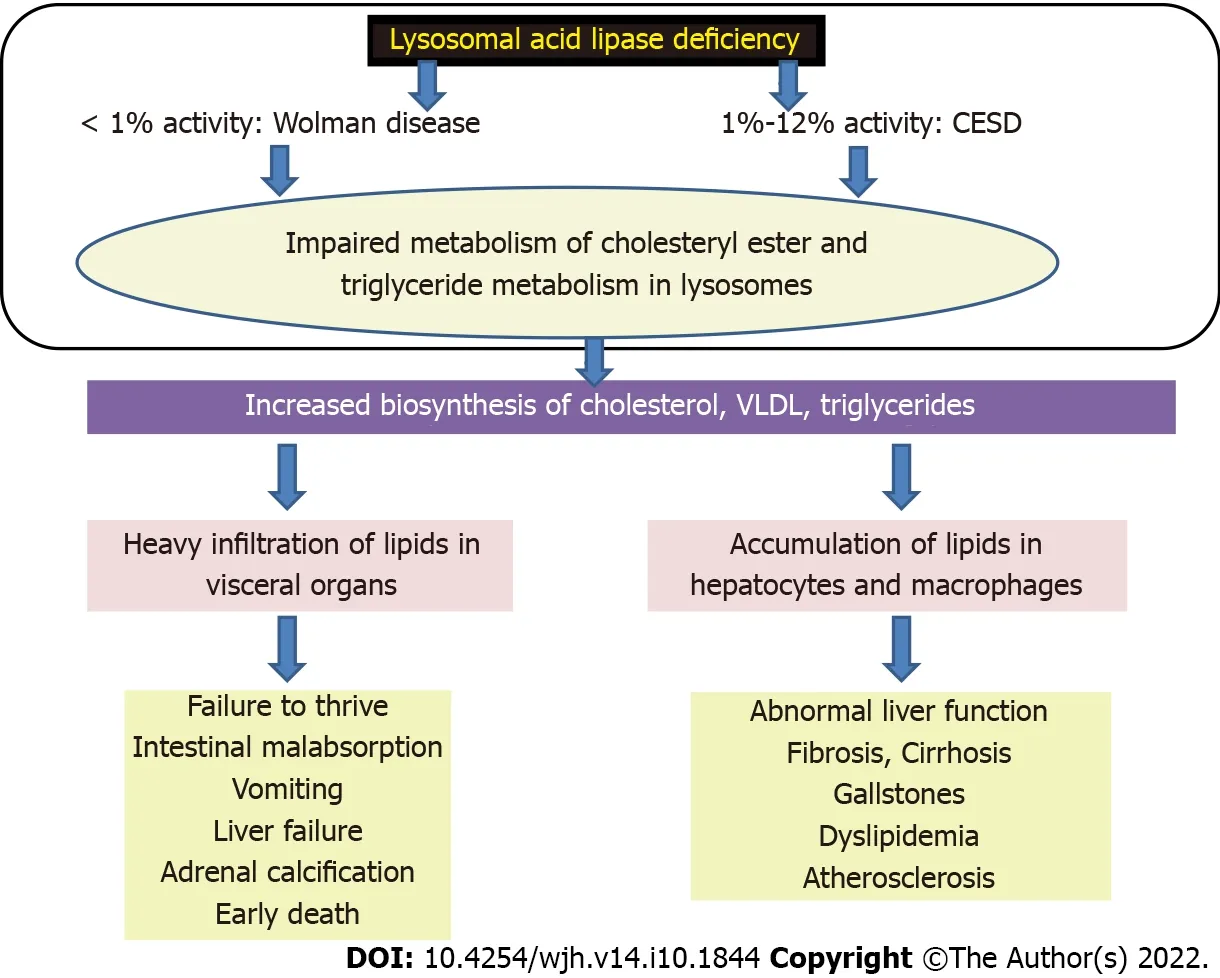
Figure 7 Pathogenesis of lysosomal acid lipase deficiency. CESD: Cholesteryl ester storage disease; VLDL: Very low-density lipoproteins.
CESD
Clinical features and natural history in CESD
In CESD, residual lysosomal activity is 1%-12%[65]. This is a heterogeneous disorder with variable age of presentation from infancy to adulthood. Disease manifestation is also variable, which includes failure to thrive, vomiting, diarrhea, asymptomatic hepatomegaly, premature atherosclerosis, and cirrhosis. CESD patients often have dyslipidemia in the form of high total cholesterol, low-density lipoprotein (LDL), triglycerides, and low high-density lipoprotein (HDL). Adrenal calcification is usually seen in WD but it can be present rarely in CESD patients. CESD patients usually survive till adulthood.
Clinical settings to suspect CESD
CESD should be suspected in non-obese patients with hepatomegaly and unexplained hypertransaminasemia with abnormal lipid profile (low HDL and high LDL)[65,66].
The clinical situations to suspect CESD are: (1) Lean nonalcoholic fatty liver disease (NAFLD); (2) obese patients with persistent hepatomegaly, elevated transaminases, and abnormal lipid profile not responding to effective body mass index reduction; (3) adolescent and young adults diagnosed with NAFLD but liver biopsy showing microvesicular steatosis; (4) NALFD in a child less than 5 years of age; (5) pediatric cryptogenic cirrhosis; (6) unexplained liver failure in a young child with hepatomegaly or fatty liver; (7) abnormal lipid profile in children without familial dyslipidemias or obesity; and (8) earlyonset gall stones or family history of cholecystectomies at a young age.
There is considerable overlap in the features of CESD and NAFLD. Obesity is associated with nonalcoholic steatohepatitis. LAL-D should be ruled out in those obese patients whose lipid profile continue to be deranged despite losing weight on treatment[65]. Microvesicular steatosis on liver biopsy may be mistaken as NAFLD[66]. NAFLD is rare in children < 3 years old, and uncommon at < 10 years of age. Unlike LAL-D, adolescents and adults with NAFLD usually have insulin resistance and hyperglycemia. Hepatomegaly is mild or absent in NAFLD as compared to marked involvement in CESD. Conversely body mass index (BMI) is high (overweight or obesity) in NAFLD as compared to near normal BMI in CESD. In lipid profile, triglycerides are mildly to moderately increased in NAFLD but near normal in CESD. HDL and LDL are mildly deranged in NAFLD as compared to moderate or marked derangement in CESD. Liver biopsy distinguishes macrovesicular steatosis in NAFLD from microvesicular steatosis in CESD[66]. Approximately 5%-15% of pediatric cirrhosis cases are cryptogenic. Screening for LAL-D is recommended in cryptogenic cirrhosis in children and adults. In those with microvesicular or mixed steatosis where Wilson disease is being considered, LAL-D should also be kept in mind. LAL-D should also be considered in unexplained liver failure in early childhood. Another setting for LAL-D is nonfamilial dyslipidemia resistant to regular treatment. The characteristic lipid profile in these patients is LDL > 130 mg/dL and/ or HDL < 40 mg/dL. These patients are non-obese and had normal fat distribution and normal fasting glucose[67,68]. Biochemical liver abnormalities are present early in the course of LAL-D disease. Low ALT, AST, and albumin with elevated GGT and bilirubin levels are characteristics in the early-onset form which is more aggressive in nature[66].
There is a genotype-hepatic phenotype correlation in CESD. Of the 32 known CESD mutations, 50% are missense, 25% are small deletions/insertions, 16% are non-sense, 6% are consensus splice-site mutations, and 3% have large deletions. The most common mutation, E8SJM-1G>A, has been found only in CESD patients. In CESD and WD, nonsense, small deletions/insertions, splicing, and missense mutations can be found. LIPA mutations that encode mutant enzymes with residual activity are found in patients with CESD[69]. Individuals of Jewish ancestry (allele frequency of 1 in 32) have the LIPA founder mutation G87V (also described as G66V)[68]. E8SJM-1G>Ahomozygotes have limited genotypephenotype correlation with diversity in presentation and progression. Almost all E8SJM-1G>Ahomozygotes in the patients have onset of symptoms in thefirst years of life, the majority by 6 years of age. Liver diseases among the reported E8SJM-1G>Ahomozygotes range from microvesicular steatosis tofibrosis andfibrosis with septal bridging, indicative of cirrhosis. Progression of the liver disease occurs later in adult life. Gastrointestinal involvements among E8SJM-1G>Ahomozygotes are gastrointestinal lipid accumulation, severe, acute, and chronic diarrhea, malabsorption, abdominal pain, and perforated gastric ulcer. Other extrahepatic manifestations are growth failure, anemia, respiratory infections, and coronary artery disease. Disease progression is variable. It is rapid in some patients and slow in others. Ultimate consequence is hepaticfibrosis and complications of atherosclerosis[70]. Several patients are compound heterozygotes for the H129P (histidine to proline) missense mutation (4.6% of normal enzyme activity) and the common E8SJM-1G>Aallele (genotype H129P/E8SJM-1G>A). This group has adultonset cirrhosis, portal hypertension, and liver failure by the 4-5thdecades[71]. CESD patients with the T288I/T288I (3.6% of normal LAL activity) and G342R/S289C genotypes have a WD-like presentation (infantile-onset, diarrhea, adrenal calcifications, and failure to thrive). However, they have sufficient residual LAL activity to survive into the second or third decades of life and also after LT[72]. Patients homozygous for H295Y (2.9% of normal LAL activity) also have infantile-onset CESD, requiring LT in the second decade[71]. Hence, early onset of disease manifestations may rapidly progress in childhood or adolescence. Patients with slower variant are usually healthy till adulthood when liver failure sets in, resulting in LT or death[73].
Burton et al[74] described 32 children with the progression of LAL-D over 13.3 (1.8-38.8) years; 25% of children were aged < 12 and 12-18 years, while 50% were > 18years. The patients had a high frequency of hepatomegaly (84%) and splenomegaly (88%). ALT (89.2 ± 42.1 U/L) and LDL (194.1 ± 63.1 mg/dL) levels were elevated. Age at onset, age at starting antilipidemic therapy, age at first recorded evidence of fibrosis or cirrhosis, and age at LT were 5.8 (0.0-42.0), 9.2 (2.0-43.2), 9.2 (1.9-41.0), and 13.0 (5.8-43.5) years, respectively. The authors concluded that the median time to an event was approximately 3.1 years. Bernstein et al[75] reviewed 135 patients with CESD. Age at onset in 35 (27%) severely affected children was between birth and two years, 81 (62%) presented between age 3 and 12 years, and 15 (11%) had an adolescent onset disease. Hepatomegaly and splenomegaly were present in 99.3% and 74%, respectively. Pathologic liver biopsy was reported in 83%, pathognomonic crystals/clefts in 16%, reduced LAL activity in 83%, and mutational diagnosis in 41%. AST and ALT levels were 54 (9-5240) and 52 (15-2340) U/L, respectively. Esophageal varices were reported in 12 patients, including nine from 5 to 20 years of age. Of the 11 reported deaths, 73% were due to liver failure between 7 to 56 years of age. Half of the deaths were under 21 years of age. Two cases of HCC were reported at the ages of 11 and 52 years. Adrenal calcifications were present in nine CESD patients aged < 1 to 10 years.
Pathology in CESD
The liver appears orange-yellow in color on gross examination. Microvesicular steatosis involving hepatocytes, Kupffer cells, and macrophages occurs due to massive lysosomal accumulation of CE and triglycerides. This progresses tofibrosis, and further into micronodular cirrhosis. On light microscopy, there is diffuse, uniform microvesicular steatosis with minimal zonal differences within the hepatic lobule. Foamy macrophages containing lipids and ceroids are present in the sinusoids and portal tracts. In contrast to macrophages, ceroid accumulation does not accompany lysosomal lipid accumulation in hepatocytes. Specific immunostains for the lysosomal lipid accumulation are LAMP1, LAMP2, LIMP2, and cathepsin D[76]. Pathognomonic birefringent CE crystals are observed in hepatocytes and/or Kupffer cells under polarized light. Fifty-eight percent of patients have specifically described birefringent, needle-shaped CE crystals, and 23% additionally cases have CE hepatocyte accumulation. CE deposition may be found in 80% by either frozen biopsy, polarization microscopy, or electron microscopy. Fixed paraffin-embedded sections show remnant clefts where the lipid had been extracted during dehydrating procedures. These crystals and clefts are seen by electron microscopy. They are limited by a single lysosomal membrane or appear free in the cytoplasm. Sixty-four percent of cases havefibrosis and/or cirrhosis. Among this group, sinusoidal, periportal, or septalfibrosis is seen in 50%, cirrhosis in 29%, and hepatocyte necrosis is reported in 7% of patients[76]. There is evidence that the disease is seen in utero. Fetal hepatocytes and syncytiotrophoblasts of the chorionic villi show marked membrane-bound CE accumulation and cholesterol infiltration. Necrosis of enlarged fetal adrenal glands is reported[77]. Gastrointestinal lipid and CE accumulate in the villi of the lamina propria, smooth muscle, vascular pericytes, and lacteal endothelium. Foamy macrophages are seen in the bowel mucosa[75].
Therapy and LT in CESD
ERT in CESD is a viable option. Sebelipase alfa is a recombinant human LAL that is expressed in egg whites from transgenic hen oviduct cells. ERT has considerable success in the late-onset LAL-D[76]. In a trial (LAL-CL01), nine patients were treated with four once-weekly intravenous infusions of sebelipase alfa at a dose of 0.35 mg/kg, 1 mg/kg, or 3 mg/kg[74]. After a median washout period of 15 wk, these patients entered another extension trial (LAL-CL04) with the same dose being continued for another 4 wk before transitioning to infusions (1 mg/kg or 3 mg/kg) every other week for a total of 12 wk. In the seven patients with completed 12 wk therapy, reductions in mean AST and ALT concentrations (P ≤ 0.05), triglycerides (P = 0.016), total cholesterol (P = 0.047), and LDL (P = 0.078), and increases in HDL (P = 0.016) were appreciated from baseline[78]. This cohort was further treated with infusions every other week for 52 wk, demonstrating the long-term efficacy of this treatment. None of the patients developed autoimmunity and maintained favorable liver functions and lipid profiles[79]. In a multicenter randomized phase 3, placebo controlled trial named ARISE, Burton and colleagues showed the effectiveness of sebelipase alfa in 66 adults and children (NCT01757184; 24 patients were aged < 12 years). A dose of 1 mg/kg was infused every other week for 20 wk followed by an open-label period for another 16 wk during which both groups received treatment. Primary outcome of normalization of ALT levels at 20 wk (31% vs 7%; P = 0.03) was better in the treatment group. Favorable changes from baseline were also seen in the treatment group as compared to placebo regarding LDL cholesterol (P < 0.001), non-HDL cholesterol (P < 0.001), triglycerides (P = 0.04), HDL cholesterol (P < 0.001), and AST (P < 0.001). The treatment group also had lesser hepatic fat content (P < 0.001), steatosis (P = 0.42), and reduction in spleen volumes (P < 0.001). Liver volume change was not significantly different between the two groups[80]. Subsequently, all the patients entered a 130 wk, open-label extension period, and a 104 wk, openlabel expanded treatment period. Age at randomization was 13 (4.7-59) years. Patients who crossed over from placebo to ERT showed improvements in liver enzymes that were similar to the ERT group in the previous double-blind trial. Thirteen patients had infusion-associated reactions and six developed antidrug antibodies[81].
Bernstein et al[82] described 18 childhood-onset LAL-D post-LT. LT was performed for progressive liver dysfunction without ERT pre- or post-LT. Despite LT, extrahepatic progression occurred in 11 patients (61%) and death in six (33%). Liver allograft and post-mortem liver biopsies showed histological recurrence. Hence, it was concluded that LT is required in LAL-D-associated liver failure, but LT cannot prevent disease progression and recurrence. The pathophysiology is predominantly mediated by deficient enzyme activity in bone marrow-derived monocyte-macrophages. Bernstein et al[82] also reported a review of cases where LT had been performed in children aged 5 to 14 years. Six post-LT patients were followed from 10 to 36 mo. Except one with an incidental HCC, the rest did not have any complications. One patient who had an LT atfive years of age developed rejection and congestive heart failure. In two patients with more thanfive years of follow-up, one 14-year-old child developed end-stage renal failure due to glomerular sclerosis, tubular atrophy, and interstitialfibrosis. The patient had extensive vascular lipid accumulation resulting in atherosclerosis. The lipid deposition in the renal vascular system signifies systemic lysosomal CE accumulation despite LT[82].
WD
Of the 19 mutations in WD, 37% are small deletions or insertions, 26% non-sense, 21% consensus splicesite mutations, 10% missense lesions, and 5% a large deletion. The two exon 8 splice-junction variants, E8SJM+1G>Aand E8SJM+3C>T, occur only in WD patients. Most severe LIPA gene mutations results in markedly reduced or no LAL activity in patients with WD[69]. Patients present just after birth, most commonly at 2-4 mo of age. Heavy accumulation of cholesteryl esters and triglycerides in visceral organs is a process that starts in utero. The features are adrenal necrosis, polyhydramnios, and microvesicular steatosis[83]. Adrenal infiltration leading to necrosis in the fetal stage leads to adrenal calcification described in about 50% of infants born with the condition (Figure 8). Infants with WD present with profound failure to thrive, hepatosplenomegaly, vomiting, and liver failure. Chronic diarrhea or steatorrhea due to the disease process itself and severe malabsorption are an important feature. Hence, the triad of WD is intestinal malabsorption, liver failure, and adrenal insufficiency[83]. Jones et al[84] showed that the median age at death was around 3.7 mo. In the untreated, the survival beyond 12 mo was highly unlikely [estimated probability 0.114 (95%CI: 0.009-0.220)]. Among the patients with evidence of early growth failure, the median age at death was 3.5 mo with even lower estimated probability of survival at 12 mo [0.038 (95%CI: 0.000-0.112)]. Despite hematopoietic stem cell transplant (n = 9) or LT (n = 1), survival was still poor (median age at death, 8.6 mo). Two open-label studies of ERT with sebelipase alfa were conducted in infants with WD. The VITAL study consisted of infants treated with once-weekly intravenous infusions of sebelipase alfa with a phase 2 dose-escalation study [LAL-CL08 (CL08)]. The analysis population contained 19 patients (9 in VITAL; 10 in CL08). Kaplan–Meier estimates of survival at 12 mo and 5 years of age were 79% and 68%, respectively. Overall, the median age of surviving patients was 5.2 years in VITAL and 3.2 years in CL08. Decreases in hepatosplenomegaly were noted in both studies. Short-term transfusion-free periods were seen in 100% of patients in the VITAL study for a period of 4.6 (0.3-16.6) mo and 70% in the CL08 study for 5.5 (3.7-19.6) mo. None of the patients discontinued therapy. Most infusion-associated reactions (94% in VITAL and 88% in CL08) were mild or moderate in severity[84,85].
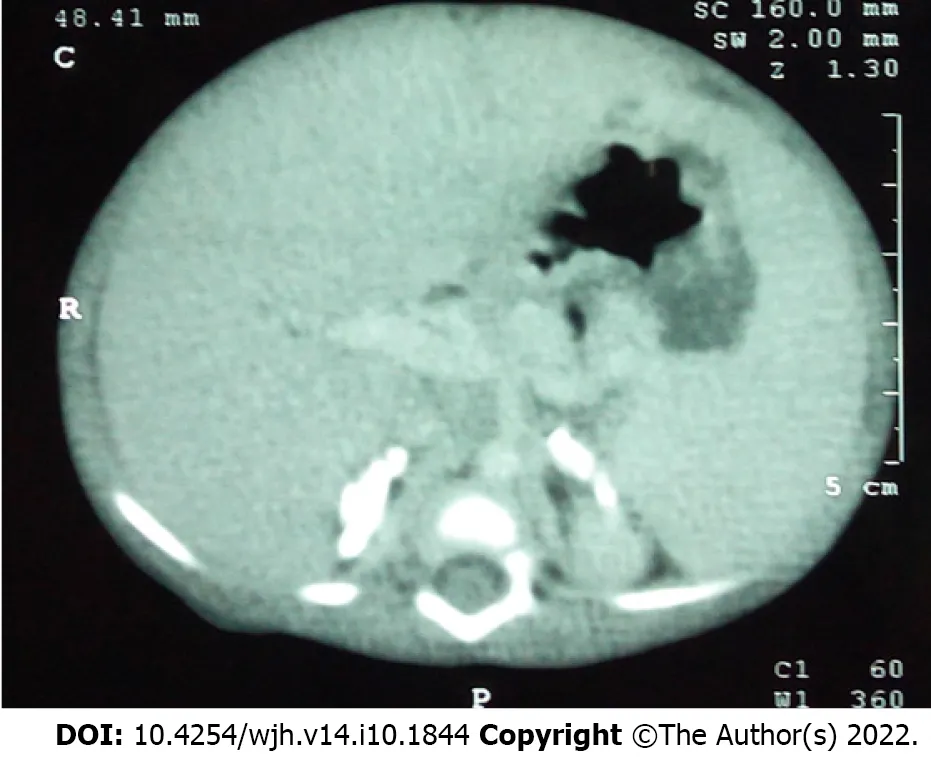
Figure 8 Computed tomography of the abdomen in an infant with Wolman disease showing bilateral adrenal calcifications.
OTHER LSDS WITH LIVER DYSFUNCTION
Asymptomatic hepatomegaly is a common component of LSDs but liver dysfunction is not a usual presentation. Few reports suggest that mucopolysaccharidosis type VII (MPS, Sly syndrome) may rarely present with neonatal cholestasis, which may lead to progressive worsening and death. Gillet et al[86] diagnosed MPS type VII in a newborn with coarse facies and neonatal cholestasis. The diagnosis was made based on high urinary glycosaminoglycans, Alder-Reilly granules within the granulocytes, and absent β-glucuronidase activity in leukocytes. In another case report, a 55-d-old baby with cholestatic jaundice and coarse facies was diagnosed with MPS type VII on genetic analysis. The patient died at 7 mo of age due to progressive liver disease[87]. Farber’s disease type IV has also been reported to present with neonatal cholestasis. Willis et al[88] reported two siblings born of nonconsanguineous parents, presenting with jaundice in the early neonatal period with rapid progression of liver disease and death at 32 and 52 d, respectively. The diagnosis was made on liver biopsy in which electron microscopy showed lysosomes containing curvilinear tubular bodies or Farber bodies. Many other LSDs are associated with hepatosplenomegaly in the newborn period, such as sialidosis, galactosialidosis, multiple sulfatase deficiency, I-cell disease, infantile sialic acid storage disease, and prosaposin deficiency[89-92]. Hochman et al[93] described a 9-d-old baby with mild jaundice who developed hepatosplenomegaly by 1 mo of age. This was later concluded as bile duct involvement in I-cell disease. One of the mechanisms of infantile hydrops is due to hypoproteinemia caused by liver dysfunction. LSDs associated with congenital ascites have been reported with sialidosis type II, galactosialidosis, isolated sphenoid sinus disease, Salla disease, MPS types IV and VII, GM1 gangliosidosis, I-cell disease, and Farber disease[94,95]. In cases of hydrops, demonstration of highly vacuolated storage cells in proband placental histology can serve as an early diagnostic clue for enzymatic testing in the further pregnancies[80,96].
CONCLUSION
Liver dysfunction in LSD poses a great challenge for pediatric and adult hepatologists. GD, NPD, and LAL-D are the most important LSD that has liver dysfunction. The hepatologist needs to have a high degree of suspicion to differentiate LSDs from other liver diseases. Extrahepatic involvement is the clue to the bedside diagnosis. Unexplained organomegaly, portal hypertension, and fatty liver are important presentations. Neonatal cholestasis and ascites are rare presentations in infants. Those presenting with neonatal or early onset of liver disease have a universally poor prognosis. Diagnosis is mainly dependent on tissue, enzyme activity, and genetics. If available, specific ERT and SRT should be conducted before irreversible organ damage occurs. Vigilance for progression has a key role in management. Those with progressive liver disease require LT. However, extrahepatic progression of the disease is often noted. Future research should preferably focus on long-term data with enzyme replacement, drug chaperone therapy, and gene therapy.
ACKNOWLEDGEMENTS
Dr. Ruchi Gupta, Department of Hematology and Dr. Rachna Agarwal, Department of Ophthalmology, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Raebareli Road, Lucknow, 226014, Uttar Pradesh, India, contributed to the manuscript.
FOOTNOTES
Author contributions: Sen Sarma M contributed to the conception and final drafting of the manuscript; Tripathi PR contributed to the data collation and primary drafting of the manuscript.
Conflict-of-interest statement: No conflict of interests.
Open-Access: This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin: India
ORCID number: Moinak Sen Sarma 0000-0003-2015-4069; Parijat Ram Tripathi 0000-0002-2690-3641.
S-Editor: Zhang H
L-Editor: Ma JY-MedE
P-Editor: Zhang H
杂志排行

World Journal of Hepatology的其它文章
- Immunotherapy for hepatocellular carcinoma: A promising therapeutic option for advanced disease
- Long-term and non-invasive in vivo tracking of DiD dye-labeled human hepatic progenitors in chronic liver disease models
- Quality of life, depression and anxiety in potential living liver donors for pediatric recipients: A retrospective single center experience
- Hepatic involvement in children with acute bronchiolitis