整合生物信息学分析痤疮的关键基因与免疫浸润水平
2022-11-28樊文龙王红心陈东宇杨晓雨黄巧胡敏潘素跃王朴何玉清
樊文龙,王红心,陈东宇,杨晓雨,黄巧,胡敏,潘素跃,王朴,何玉清,2
1.广东医科大学公共卫生学院流行病与卫生统计学系,广东医科大学医学系统生物学研究所,广东 东莞 523808;2.东莞市寮步医院,广东 东莞 523400
痤疮作为一种生活中常见的慢性炎症性皮肤病,在不同人群中均有发生,好发于青春期[1],在面部、胸背部较为常见,表现为粉刺、丘疹、脓疱、结节、囊肿甚至瘢痕等不同程度的损害。2019年,全球痤疮发病人数约为1.17亿例,1990—2019年期间,全球痤疮的整体患病率和疾病负担均逐年升高[2]。痤疮可能会影响患者生活质量[3-5]。痤疮的发生受多种因素影响,主要包括遗传、雄激素刺激皮脂腺异常角化、微生物影响,炎症的病理免疫反应等[6]。雄激素/雄激素样受体、Toll样受体、表皮生长因子受体、过氧化物酶体增殖物激活受体、成纤维细胞生长因子受体和FOXO1转录因子等均参与寻常痤疮的发病过程[7],但其发病机制仍不完全明晰。近年来,利用生物信息学的方法进行数据挖掘已然成为一项探究发病机制的研究手段。因此,本研究通过生物信息学方法对从GEO数据库获取的数据进行分析,探究与痤疮发病相关的关键基因,为后续相关研究提供理论参考。
1 材料与方法
1.1 数据来源
从GEO数据库(https://www.ncbi.nlm.nih.gov/geo/)中下载三个数据集:GSE108110(GPL570平台)、GSE53795(GPL570平台)和GSE6475(GPL571平台);其中GSE108110更新于2020年3月;GSE53795更新于2019年3月;GSE6475更新于2018年12月。这三个数据集的样本均为痤疮患者皮损组织和正常组织,其中GSE108110皮损组织和正常组织各18例,GSE53795皮损组织和正常组织各12例,GSE6475皮损组织和正常组织各6例,共纳入72例样品组织。
1.2 差异表达基因(differentially expressed genes, DEGs)的提取
利用Rstudio对下载好的数据进行处理,并安装“impute” R包对缺失值采用最近邻居法(K-nearest neighbor, KNN)进行填充,安装 “limma” R包对处理好的基因进行分析,在校正后P<0.05,|logFC|>1的标准下,筛选出痤疮的DEGs,绘制韦恩图筛选出三个数据集的共同DEGs。使用在线分析软件平台SangerBox(http://vip.sangerbox.com/),去除三个数据集的批次效应并合并数据,用于后续分析,该平台整集100多种常用的分析方法,涵盖统计、分析、可视化等工具,提供了大量广泛使用的生物信息学分析工具[8]。
1.3 加权基因共表达网络分析(weighted gene co-expression network analysis, WGCNA )[9]
根据标准化后的基因表达矩阵,筛选出皮损组和对照组有差异的基因(P<0.05),利用SangerBox构建WGCNA,计算软阈值,进行基因聚类和模块划分(最小模块基因数取30,敏感性取3,模块合并阈值取0.25),评价基因模块和痤疮表型的关联性,筛选出正相关和负相关最高的模块。
1.4 基因功能和通路富集分析
对两基因模块中的基因和共同DEGs取交集,使用SangerBox对交集基因进行基因本体论(gene ontology, GO)功能注释,同时对交集基因进行京都基因与基因组百科全书(kyoto encyclopedia of genes and genomes, KEGG)通路富集分析,在P<0.05的条件下,筛选显著的功能和通路。
1.5 蛋白质互作网络(protein-protein interaction network, PPI network)的构建与分析
使用STRING在线工具(https://string-db.org/)分析蛋白质-蛋白质相互作用,并使用Cytoscape(version:3.8.2)进行可视化,构建PPI网络[10-11];使用CytoHubba插件中的4种常用算法(MCC、Degree、Betweenness和Closeness)分别筛选出PPI网络中前20的关键基因,并取它们的交集。
1.6 免疫细胞浸润分析
基于SangerBox在线分析软件,通过CIBERSORT算法进一步研究痤疮样本中22种免疫细胞浸润的差异[12],评估22种免疫细胞浸润的比例,分析关键基因与免疫细胞浸润的相关性。
2 结果
2.1 差异表达基因提取
数据处理后,在校正后P<0.05,|logFC|>1的条件下,GSE108110、GSE53795和GSE6475数据集中分别筛出547、1 106和284个DEGs,其中GSE108110上调基因472个,下调基因75个;GSE53795上调基因981个,下调基因125个;GSE6475上调基因261个,下调基因23个。筛选三个数据集的共同DEGs,共筛选出154个DEGs,其中上调基因152个,下调基因2个(表1,图1)。
2.2 WGCNA
WGCNA分析结果显示,软阈值β为24,signed R2为0.85,平均连接度为3.41,网络符合无标度网络的分布。在最小模块基因数为30、敏感性为3、模块合并阈值为0.25的标准下,基因集被分为了4个共表达模块,其中lightcyan模块(4 419个基因)与样本表型显著正相关(r=0.92,P<0.001),green模块(228个基因)与样本表型显著负相关(r=-0.65,P<0.001,图2)。
2.3 基因功能和通路富集
对WGCNA中正、负相关性最高的两模块进行功能富集分析,GO富集结果显示,在生物过程方面,主要集中在免疫系统过程、免疫反应、细胞激活等;在细胞成分方面,基因主要集中于质膜部分、囊泡、细胞内囊泡等;在分子功能部分,基因主要与信号受体结合、含有蛋白质的复合体结合、催化活性等有关(图3A);KEGG结果显示,基因主要集中在细胞因子-细胞因子受体的相互作用、趋化因子信号传导、NF-κB信号传导等途径(图3B)。对这154个DEGs进行功能富集分析,GO富集结果显示,在生物过程方面,主要集中在免疫系统过程、免疫反应、细胞激活等;在细胞成分方面,基因主要集中于分泌颗粒、分泌囊泡、细胞质囊泡等;在分子功能部分,基因主要与信号受体结合、细胞因子活性、趋化因子受体结合等有关(图3C);KEGG结果显示,基因主要集中在病毒蛋白与细胞因子和细胞因子受体的相互作用、细胞因子-细胞因子受体的相互作用、趋化因子信号传导途径、NF-κB信号传导途径、IL-17信号传导等途径(图3D)。将WGCNA两模块基因与DEGs取交集,结果显示,交集基因为154个(图3E)。
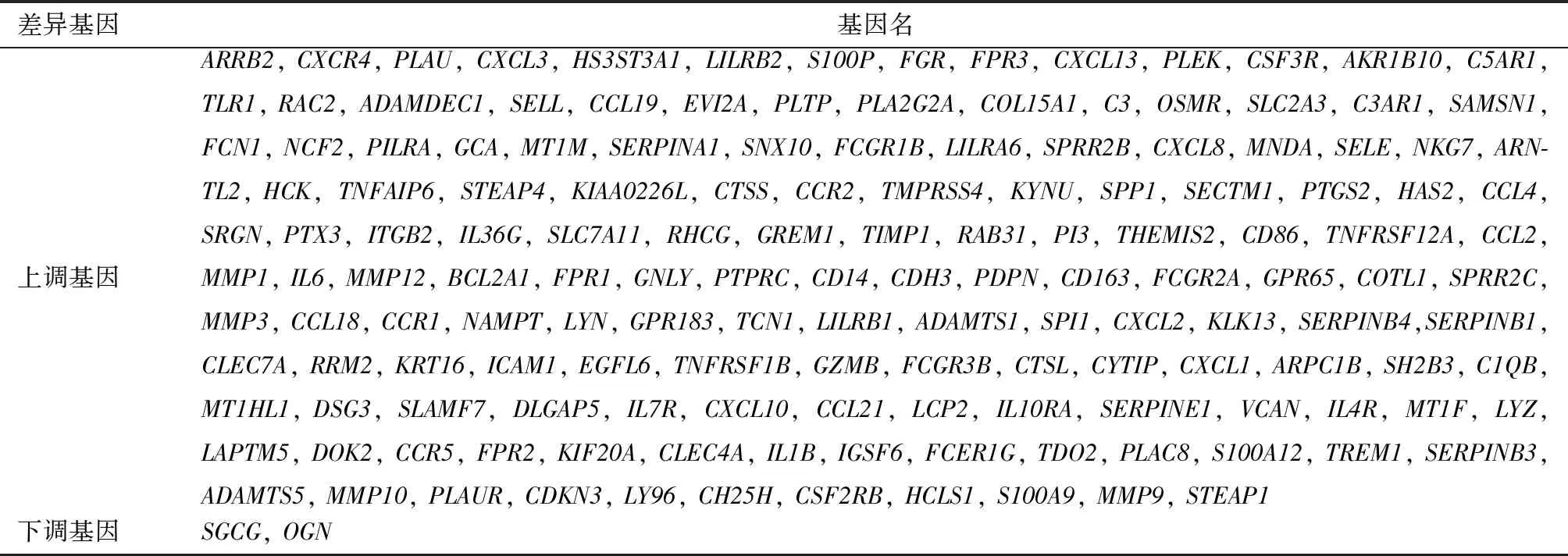
表1 共同差异表达基因Table 1 Differentially expressed genes
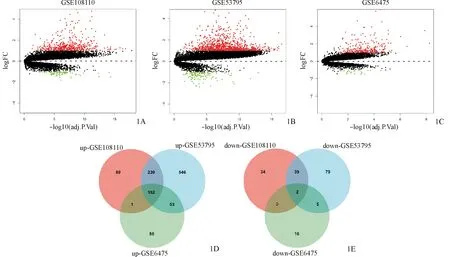
图1 差异表达基因 1A、1B、1C:三个数据集的火山图,红色代表上调基因,绿色代表下调基因,黑色代表没有显著性差异的基因;1D:三个数据集上调基因的韦恩图;1E:三个数据集下调基因的韦恩图
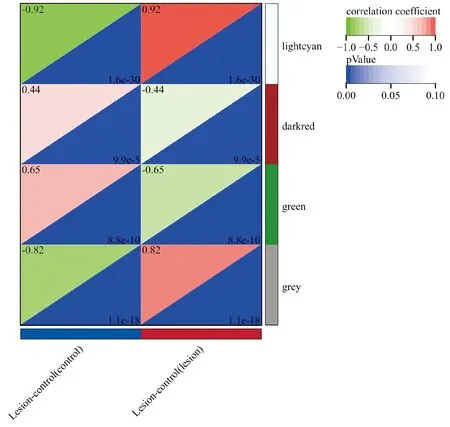
图2 各模块与痤疮的相关性Figure 2 Correlation of each module with acne.
2.4 蛋白质互作网络
使用Cytoscape构建PPI网络,得到139个节点,1 597条边,提取连接度排名前10的节点,分别是IL-6、CXCL8、IL-1B、PTPRC、CD86、MMP9、ITGB2、CCR5、CCL2、TLR1(图4A)。调用CytoHubba插件,采用4种算法筛选关键基因,提取4种算法的前20关键基因取交集,最终交集到2个关键基因,分别为CXCL8、CCR5(图4B)。
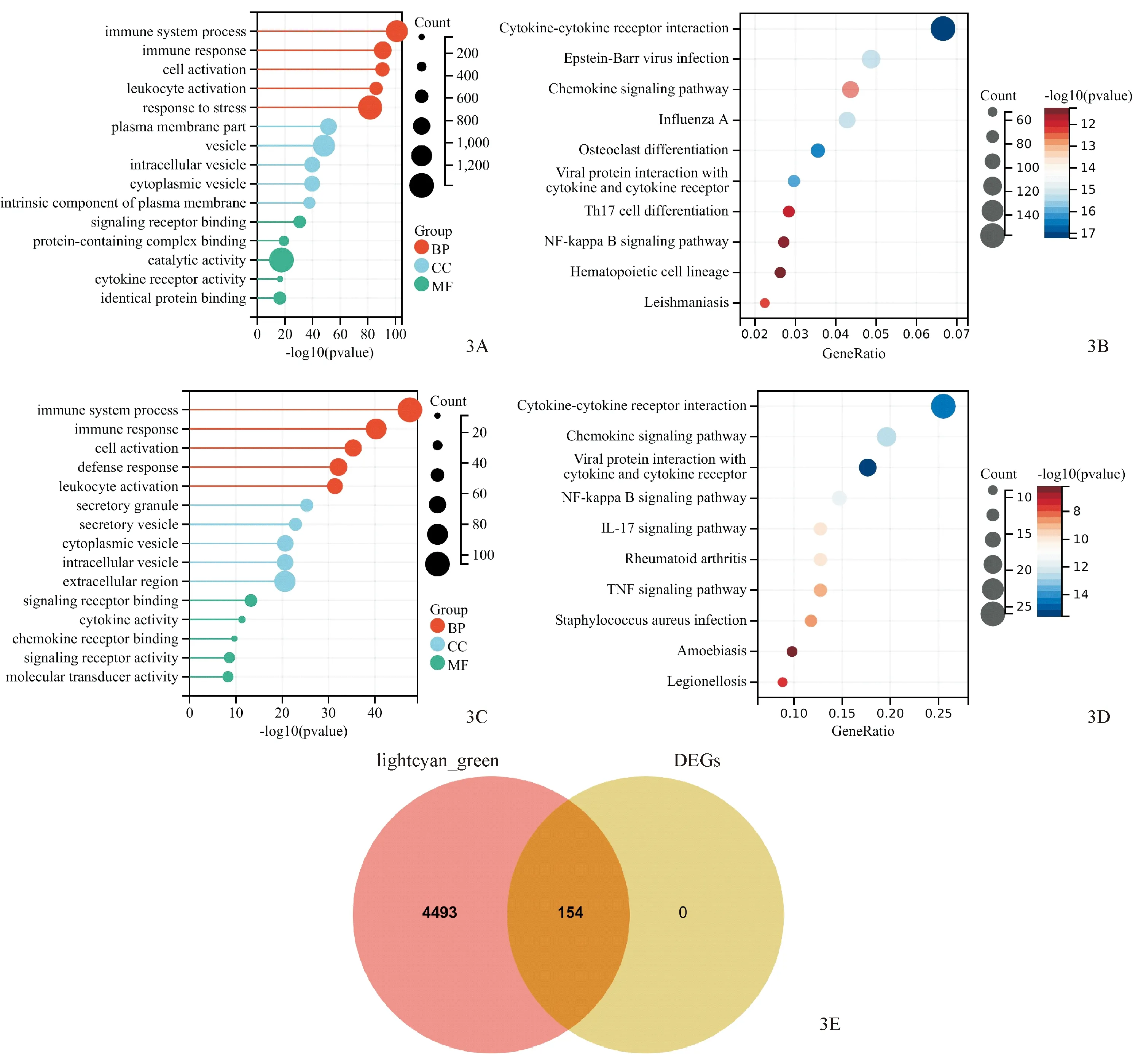
图3 基因功能和通路富集 3A、3B:两模块的GO富集和KEGG富集;3C、3D:DEGs的GO富集和KEGG富集;3E:两模块与DEGs的交集
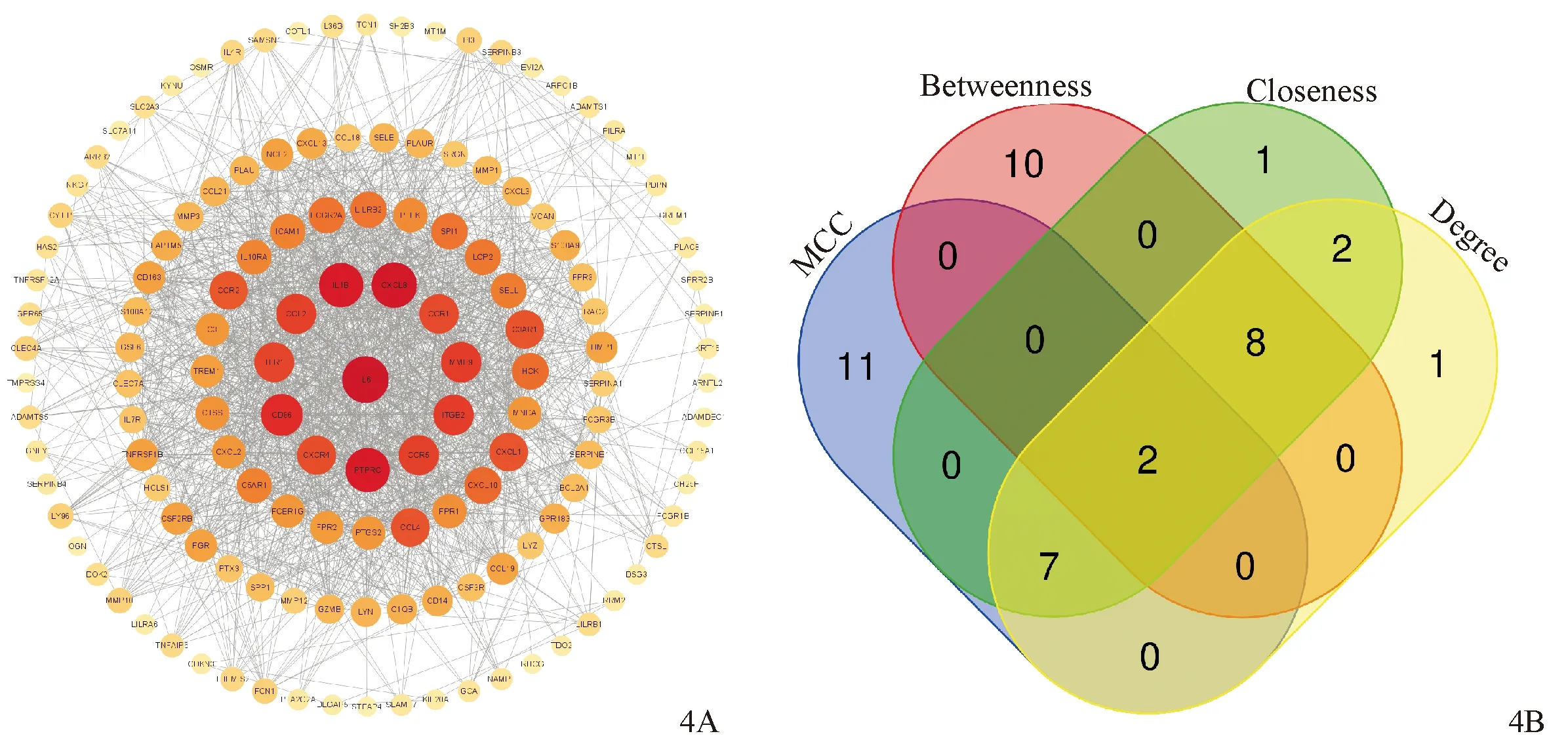
图4 PPI网络(4A)及四种算法关键基因的交集(4B)
2.5 免疫细胞浸润分析
使用CIBERSORT算法分析皮损组织与正常组织之间的差异,图5显示了两组之间22 种免疫细胞类型的一般分布,与正常组织相比,皮损组织有更多成熟B细胞、静息的CD4记忆T细胞、活化的CD4记忆T细胞、单核细胞、巨噬细胞M0、活化的树突状细胞、激活的肥大细胞和中性粒细胞,而记忆B细胞、浆细胞、调节性T细胞、静息的树突状细胞、静息的肥大细胞相对更少。
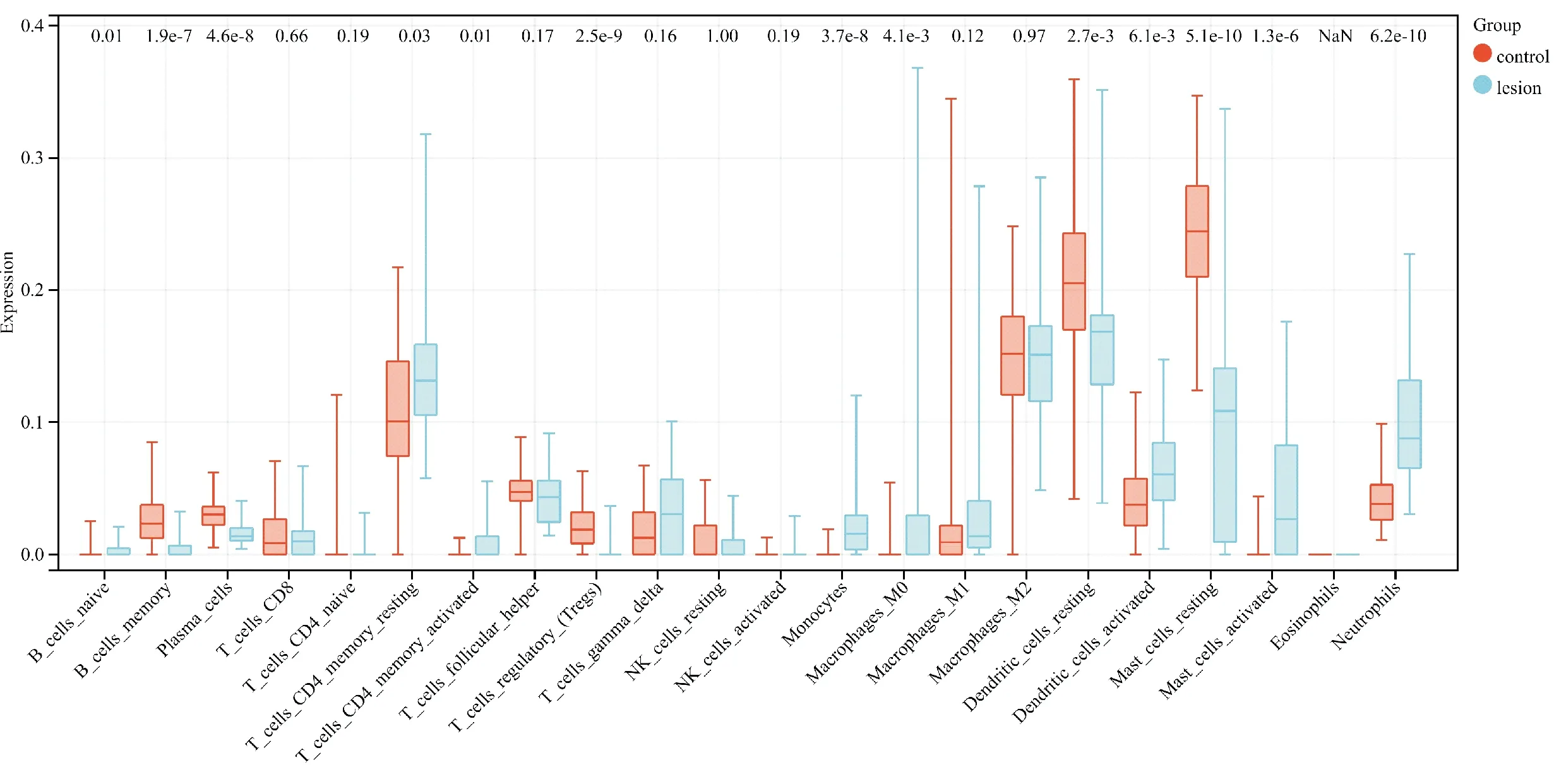
图5 22种免疫细胞在痤疮皮损组织和正常组织中浸润的分布
3 讨论
本研究采用生物信息学方法,对数据集GSE108110、GSE53795和GSE6475进行数据挖掘,以便进一步了解痤疮的发病机制。通过WGCNA筛选重要基因模块后,与差异表达基因取交集,最终得到了154个与痤疮表型相关的差异表达基因,其中上调基因数量远高于下调基因,可能原因是痤疮作为一种慢性炎症性皮肤病,痤疮发生后炎症相关基因表达,同时本研究筛选差异基因设置的标准较为严格,导致某些下调基因被过滤掉。
进行蛋白质互作分析,采用4种常用的筛选关键基因的方法取交集,最终筛选出2个hub基因,CXCL8和CCR5。CXCL8 是C-X-C基序趋化因子配体,即白细胞介素-8(IL-8),由上皮细胞和巨噬细胞等分泌。研究表明,CXCL8是肿瘤发生的关键因素,在肿瘤微环境中参与信号转导,促进肿瘤的进展[13],同时,CXCL8也参与多种皮肤病的发生相关,动物实验表明,特应性皮炎、酒渣鼻、银屑病的患者中,CXCL8均有显著增加[14-16]。另外,既往研究表明[17-18],IL-8也参与痤疮的发生发展,P.acnes通过激活Toll样受体2(TLR2)能够分泌IL-8,在SZ95皮脂细胞中,采用痤疮丙酸杆菌无细胞提取物也能够激活人NF-κB和p38 MAPK通路,并通过TLR2依赖的信号途径上调IL-8的分泌,导致炎症的发生,这与本研究中CXCL8作为上调基因的结果一致。CCR5为C-C基序趋化因子受体,在细胞趋化中发挥着重要作用,CCR5与其配体结合后,能够促进炎症反应以及诱导免疫反应中不同T细胞亚群的黏附和迁移[19]。痤疮是一种雄激素依赖性、胰岛素样生长因子IGF-1驱动的皮脂腺毛囊疾病[20],而相关文献报道,CCR5过度表达与高雄激素血症和胰岛素抵抗有关[21],CCR5的缺乏能够导致脂肪组织巨噬细胞中M2显性表型的改变,有助于减轻肥胖诱导的胰岛素抵抗[22-23]。叉头盒转录因子O1(FOXO1)作为脂肪生成的负调控因子,能够减少痤疮的发生[24],而痤疮患者体内胰岛素和胰岛素样生长因子-1的水平升高会增加Akt的活性,从而通过磷酸化降低FOXO1的核活性[25],增加痤疮的发生,本研究也显示CCR5上调,因此,推断CCR5可能通过炎症反应和胰岛素/胰岛素生长因子-1信号调节参与痤疮的发生。另外,本文KEGG富集结果显示,筛选的两个关键基因均富集于病毒蛋白与细胞因子和细胞因子受体的相互作用、细胞因子-细胞因子受体的相互作用、趋化因子信号传导途径,这两个关键基因可能通过这些通路参与痤疮的发生,而本文尚未进行实验对筛选出的基因进行验证,有待进一步实验证实。
既往文献表明,IL-17和Th17细胞与痤疮发病有关,IL-17可作为痤疮发病机制的标志物,而且可能是痤疮严重程度和瘢痕形成的潜在预后预测因子,Th17通路被激活后,可在疾病中发挥着关键作用[26-28]。Th17细胞属于CD4+T淋巴细胞亚群,CD4+T细胞在转化生长因子TGF-β与IL-21的共同诱导下分化为Th17细胞,而Th17细胞能够特异性分泌细胞因子IL-17,IL-17可刺激角质形成细胞产生促炎细胞因子、趋化因子和基质金属蛋白酶,引起炎症反应和组织破坏[29]。本研究在KEGG分析中富集到IL-17信号传导通路,在一定程度上表明IL-17在痤疮发病机制中占据重要地位,也间接表明CD4+T细胞的重要性,正如相关文献所报道,在痤疮初期,患者皮损及血清的CD4+T细胞较正常人明显增多,经过治疗后,CD4+T细胞减少,痤疮炎症病变明显改善[30-31]。本研究利用基因芯片数据集对22种免疫细胞浸润分析显示,相对于正常对照组,皮损组中有更多的中性粒细胞、活化的肥大细胞、活化的树突状细胞、活化的CD4记忆T细胞等,而调节性T细胞、静息的树突状细胞、静息的肥大细胞等相对更少,免疫细胞浸润在痤疮发生发展中的重要性不言而喻,但这些细胞具体的机制还需要进一步验证。
综上所述,本研究通过WGCNA和“limma” R包筛选出了154个差异表达基因,主要集中在病毒蛋白与细胞因子和细胞因子受体的相互作用、细胞因子-细胞因子受体的相互作用、趋化因子信号传导、NF-κB信号传导、IL-17信号传导等通路。CXCL8、CCR5这2个hub基因可能成为痤疮发病的关键基因,免疫细胞浸润水平的变化可能是痤疮发病的重要原因,在痤疮的发病中发挥着重要作用。这些结果有助于进一步阐述痤疮的发病机制,为后续的相关研究提供一定的参考。
