动物粪便中分子遗传信息的应用
2022-11-28王小琪段子渊
王小琪,段子渊
(中国科学院遗传与发育生物学研究所,北京100101)
非损伤性取样(非侵入式)是在不伤害动物的前提下,采集动物的粪便、尿液、毛发或含有口腔脱落细胞的食物残渣等可能含有遗传信息的样本(周璨林等,2015)。相比于伤害性取样,非损伤性取样方法更加简单方便,对动物机体没有伤害。在动物研究中,尤其是在野生动物的研究中使用较为广泛。其中,粪便样本因其易获得、识别性高和含有多元遗传信息的优点,极大地促进了动物学的相关研究(单磊等,2018),并由此诞生了一门新的学科:分子粪便学。目前,粪便是应用最广泛的非损伤性样本,利用粪便样本可对动物开展食性分析、肠道微生态研究、标记物筛选、种群遗传多样性分析等多方面的深入研究。尤其是近20年来,二代测序技术的出现,标志着测序正式进入到高通量时代,为遗传信息的发掘提供了大量、高效且可靠的数据,快速推动了各类组学的发展。粪便在动物研究中的传统应用方式已被多次报道(刘丙万,蒋志刚,2002;周璨林等,2015),本文就目前利用测序技术提取粪便样本中分子遗传信息的研究予以简要概述。
1 食性研究
动物的食性研究是了解动物与环境相互关系的前提。然而对于野生动物或放牧家畜来说,食物是不可控的,不了解它们吃了什么,就无法进一步研究其活动范围、饮食结构及营养代谢。除了对粪便样本的视觉观察,从20世纪80年代起,基于DNA的鉴定方法出现,到21世纪初期,DNA条形码正式出现并被标准化(Hebertet al.,2003)。DNA条形码是一个特定的短序列DNA 片段,具有物种特异性,可以被用来准确快速地识别物种。但尚未有适用于所有物种的通用条形码,为了获得更高的覆盖度和准确率,通常组合使用DNA 条形码,这些组合条形码主要来自以下基因片段:动物线粒体细胞色素C氧化酶Ⅰ基因,植物核糖体大亚基的核酮糖1,5-二磷酸羧化酶基因,叶绿体成熟酶K 基因,叶绿体trnL 内含子区和核糖体内转录间隔区(Group CPW,2009;Valentiniet al.,2009;Taberletet al.,2012;张瑞莹,2013)等。DNA宏条形码是随着二代测序技术发展而诞生的用于检测混合样本的条形码技术,可以快速、大规模地鉴定粪便样本中的DNA,确定食物范围、种类和成分。由于动物有肉食性、草食性和杂食性,所以需要对被检动物的饮食情况有所了解,才能够选择合适的引物用于DNA宏条形码检测;同时,还需要采集当地的植物或动物样本,建立适用于当地的DNA 条形码数据库,用于比对测序得到的宏条形码(刘刚等,2018)。
2 肠道微生态研究
胃肠道微生物数量众多,与宿主共同进化,保持动态平衡有助于维持宿主肠道健康(Relman,2012),也对宿主的生长、营养代谢和免疫调控起重要作用(Bäckhedet al.,2004;Kataoka,2016)。当肠道内稳态受到干扰时,胃肠道和其他器官(大脑、肝脏等)的功能会受到损害。探索肠道微生物群落结构和功能,有助于了解微生物组对宿主健康的作用、微生物组特性与宿主特性之间的关系,以及微生物组在疾病中的作用(Stulberget al.,2016)。采集粪便样本避免了肠道内取样对动物体可能造成的伤害,但在肠道微生物组研究中,有关粪便样品是否能代表肠道内容物用于肠道微生物组研究一直存在争议(Ingalaet al.,2018;Yanet al.,2019)。近期,多国科研人员共同提出了粪便微生物组倡议(Sapountziset al.,2021),并归纳了使用粪便样本进行微生物组研究的优势:(1)能可靠地代表肠道末端的微生物群落;(2)不违背动物福利及动物伦理;(3)可能含有来自瘤胃和小肠的部分微生物,这对于瘤胃和小肠内微生物的相关研究也具有意义;(4)新鲜的粪便还可以用于培养组学,分离可培养微生物;(5)可分离核苷酸或蛋白质材料,用于组学测序、诊断性PCR 和其他分子和生化研究;(6)用于鉴定非消化饲料成分的化学分析;(7)可用于体内和体外实验。
通常,绝大多数的胃肠道微生物是细菌,还有极少量的古菌、病毒、真菌和原生动物(Parkeret al.,2020)。针对微生物组的高通量测序方式常用的是:宏基因组、扩增子、宏病毒组和宏转录组。其中扩增子测序包含针对细菌和古菌的16S rRNA测序,针对真核生物的18S rRNA 测序和ITS 测序,部分常见的PCR扩增引物见表1。
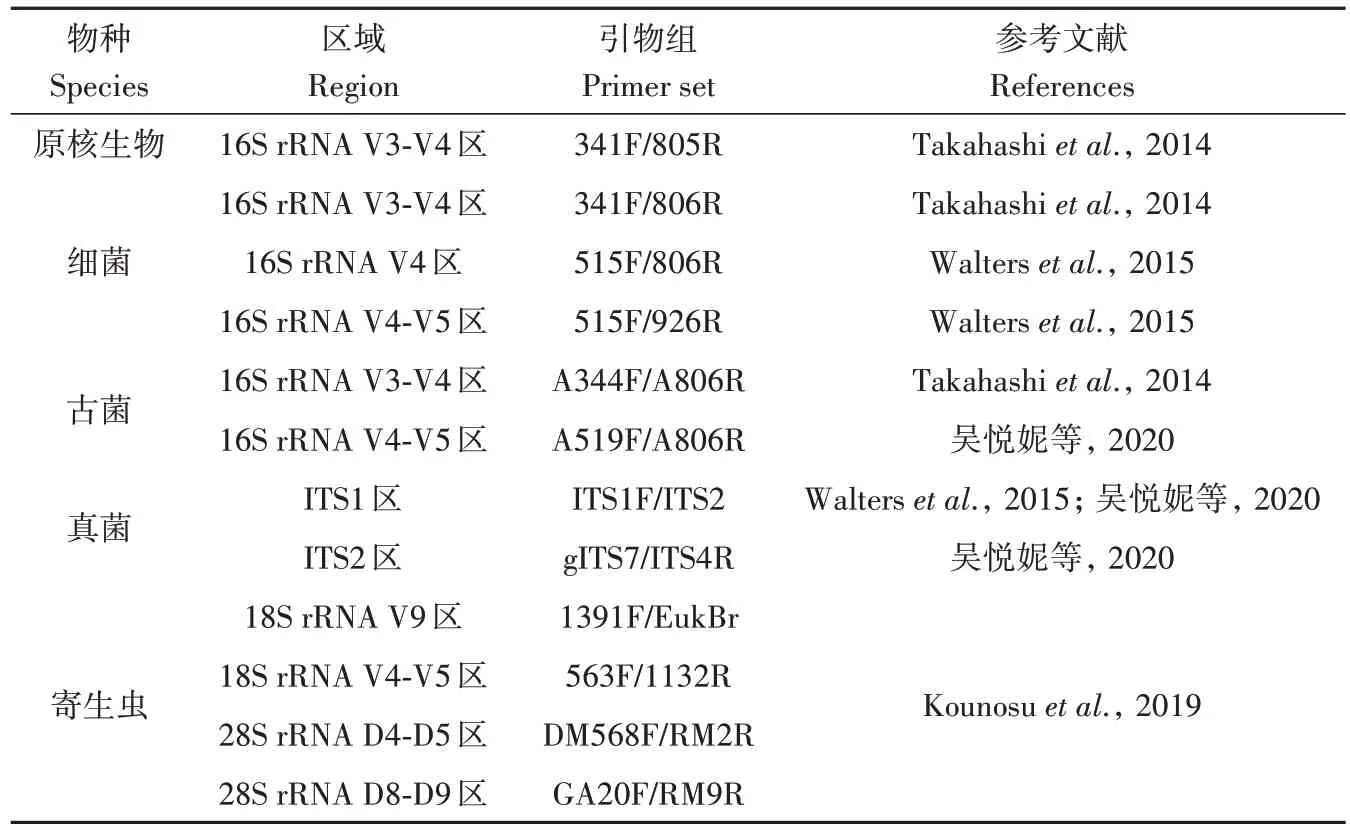
表1 用于扩增子检测的PCR扩增引物Table 1 PCR primers for amplicon sequencing
2.1 细菌研究
细菌占肠道微生物总量的90% 以上,有500~1 000 种(Kataoka,2016),被认为是肠道微生态平衡的主导者。研究表明,人类肠道细菌的稳态平衡与多种疾病,如炎症性肠病、肥胖、糖尿病、非酒精性脂肪性肝炎、慢性心脏病、癌症和自闭症等有关(Zhanget al.,2015)。目前,粪便样本在肠道细菌研究的应用上已极为成熟,对于人、家畜、实验动物和野生动物,基于扩增子(16S rRNA)测序,可以分析群落多样性,揭示细菌群落与宿主代谢、免疫的关联;利用宏基因组测序,可以解析微生物组的基因功能分布等。对于野生动物,粪便样本不仅可以帮助了解肠道细菌群落组成(Huanget al.,2020;Ishida-Kurokiet al.,2020),也可以协助鉴定肠道细菌特有的功能基因(张呈波,2021)。此外,通过宏基因组测序还可挖掘野生动物粪便细菌的功能基因,合成具有医药、食品等应用潜力的生化制品。杨金茹等(2022)从西黑冠长臂猿Nomascus concolor粪便宏基因组中筛选出的丙氨酸消旋酶基因,经扩增、重组及异源表达后,得到了稳定性更好、催化活性更高的丙氨酸消旋酶;杨正凤(2018)通过滇金丝猴Rhinopithecus bieti粪便微生物宏基因组,获得了具有优良工业特性的几丁质酶和β-半乳糖苷酶。
2.2 真菌研究
肠道真菌可能参与宿主的炎症性肠炎等疾病,但肠道内真菌群落的稳定性不如细菌群落,容易受环境因素影响(Iliev & Cadwell,2021)。由于必须满足能在37°C 下生长的条件,目前检测到能在肠道内生长和定植的真菌种类较少,主要是念珠菌属Candida(假丝酵母属)和双足囊菌科Dipo‑dascaceae(Hallen-Adams&Suhr,2017)。宏基因组测序结果显示,真菌只占人类肠道微生物的0.01%(Nashet al.,2017)。尽管真菌群落的稳定性较差,但Hoffmann等(2013)对96份粪便样本进行扩增子(ITS 区)检测,人类微生物组计划通过对317 份粪便样本的ITS区和18S rRNA基因测序(Behret al.,2018),Sun 等(2021)通过宏基因组测序对取自中国不同地域的6个民族(傣族、哈尼族、藏族、白族、苗族和汉族)的942 份粪便样本测序,发现酵母菌属Saccharomyces和念珠菌属的含量最高;其中,Sun 等(2021)还进一步确定城市化对真菌群落影响最大,会显著降低真菌丰富度。在动物肠道真菌群落研究中,通过ITS 区测序技术,粪便样本被用于评估仔猪断奶后胃肠道中真菌定植的波动情况,用以干预断奶过渡期仔猪的健康和生长状况(Arfkenet al.,2020)。苏日娜等(2022)比较了原麝Moschus moschiferus和 林麝Moschus berezovskii肠 道真菌菌群结构特性及季节因素对真菌菌群多样性的影响。
2.3 古菌研究
基于广泛使用的多相分类法(Vandammeet al.,1996),古菌不等同于细菌,被归类为一个单独的原核生物类群。尽管16S rRNA 基因序列有过于保守导致序列同源性过高的缺点,但目前依旧被认为是推断原核生物系统发育关系最重要的“标准”(Schleifer,2009)。相较于细菌和真菌,古菌有独特的遗传、生化和细胞特征。与细菌相比,古菌与真核生物有更多共同之处,包括基因组结构、转录和翻译机制等(Doolittle&Logsdon,1998)。大多数的古菌门存在于海洋、火山等环境中,对生物地化循环起着至关重要的作用(Bakeret al.,2020)。人或动物肠道中定植的古菌属仅有少数几种:甲烷短 杆 菌 属Methanobrevibacter、Methanomethylophi⁃lus属 、Methanomassiliicoccus属 、甲 烷 球 形 菌 属Methanosphaera、甲烷粒菌属Methanocorpusculum、甲烷杆菌属Methanobacterium、嗜盐古菌属Haloferax和盐红菌属Halorubrum(Chibaniet al.,2022)等。其中,广古菌门Euryarchaeota 包含广泛分布的产甲烷菌,也是研究最广泛的古菌门。尽管在猪的肠道中还存在少量其他古菌属,但丰度最高的也是与产甲烷相关的菌属(Denget al.,2021)。此外,人类肠道中的古菌含量变化与宿主的代谢、免疫有关,比如儿童哮喘、肠道功能紊乱、肥胖、结直肠癌等(Triantafyllouet al.,2014;Barnettet al.,2019;Cokeret al.,2020)。目前,对人或野生动物肠道古菌的研究,常通过采集粪便样本进行16S rRNA 测序分析,如对大熊猫Ailuropoda mela⁃noleuca、林麝粪便古菌结构组成的研究(徐谊英,2015);而经济动物(牛等)的粪便样本也可用于表征肠道古菌群落(Cendronet al.,2020)。
2.4 病毒研究
肠道病毒包括感染细菌的噬菌体和感染真核细胞的病毒,它们与细菌和肠道上皮细胞的相互作用可以激活宿主的保护性免疫途径(Iliev &Cadwell,2021)。肠道病毒检测的常规方法是有针对性地设计病毒相关引物或探针,对粪便或肛拭子中提取的病毒核酸进行荧光定量PCR 检测,成本低且灵敏度高,是目前通过人或动物粪便检测诺如病毒(钱明明等,2015)、猪δ 冠状病毒(肖帅,2018)、新型冠状病毒(吴冰珊等,2020)等单一病毒的常规方法。随着高通量测序技术的发展,了解肠道病毒的手段也发生了革命性进步,利用宏病毒组,也称为病毒宏基因组,不仅可以检测样本中所有病毒的遗传物质、确定病毒组成,还可以进行病毒溯源(Rosario&Breitbart,2011)。对粪便样本进行宏病毒组测序已经是了解野生动物粪便病毒群落结构和发现新病毒不可或缺的工具。古文鹏和代解杰(2019)探索树鼩Tupaia belangeris肠道病毒的群落结构和分布特征、文陇英等(2022)评估四川邛海湿地公园9种鸟类的病毒多样性等。
2.5 寄生虫调查
人畜共患寄生虫会对人、野生动物和家畜的生产生活造成了极大的负面影响。由于大部分寄生虫的虫卵、卵囊或幼虫可以随宿主粪便排出,检查粪便可以确定寄生虫感染的情况(谷玉,道力高,1999)。使用涂片、沉淀、漂浮和培养等传统检查方法,无法获得寄生虫的分子遗传信息,也不适用于处理较大的样本量。野生动物中常见的人畜共患寄生虫主要分为原虫和蠕虫(线虫、绦虫和吸虫)(车丽美等,1996)。以常见的人畜共患隐孢子虫Cryptosporidiumspp.、毕氏肠微孢子虫Enterocyto⁃zoon bieneusi和十二指肠贾第虫Giardia duodenalis为例,通过设计特异性引物(针对小亚基rRNA)进行巢式PCR 可以检测出来。Zhang 等(2019)利用该方法对青藏高原地区放牧的牦牛、藏绵羊和骆驼等动物的粪便样本分析了上述3 种寄生虫的流行病学情况。多项流行病学调查可发现,动物中常常出现寄生虫混合感染的情况(陈小丽,2013;李东方等,2018;李瑞琪等,2020),而扩增子等高通量测序技术可以灵敏、高效且全面地检测动物是否感染寄生虫和寄生虫的感染种类及亚型。在目前使用ITS区检测寄生虫的测序中,18S rRNA 基因的V4 和V9 区是最常用的目的序列(Hadziavdicet al.,2014)。对云南独龙牛Bos frontalis粪便样本寄生虫调查(岳凤娇,2021)、野生大鼠粪便蠕虫多样性评估(Tanakaet al.,2014)。由于ITS 区和大亚基rRNA(28S rRNA 等)的多样性高于18S rRNA,ITS 区和28S rRNA 基因也可被用于扩增子测序(Kounosuet al.,2019)。
3 生物标记物
生物标记物可应用于疾病诊断、疾病分期和分类,预后指标、干预措施的临床反应预测和监测;还可用于临床转化研究,加快药物的研发进程(Group BDW,2001)。常利用PCR、荧光定量PCR、酶联免疫吸附法(ELISA)、扩增子或宏基因组、代谢组和蛋白质组等技术筛选和鉴定生物标记物。
由于肠道微生态平衡与宿主的健康息息相关,当宿主的健康状况发生改变时,肠道内微生物稳态会严重失衡,其中丰度发生显著变化的微生物,可能是与此类疾病密切相关的微生物标记物。王小琪等(2022)基于16S rRNA 测序数据,筛选出了12 个与柴达木马腹泻相关的粪便微生物标记物。此外,还有中性粒细胞释放的乳铁蛋白和钙卫蛋白、肠道菌群产生的短链脂肪酸等,都可以通过宿主的体循环随粪便排出,当宿主出现炎症时,其含量会发生极大变化,此类代谢物也可以作为相关疾病的生物标记物。目前已有多种粪便微生物和代谢物被确定为疾病相关的标记物(表2),成为诊断或治疗疾病的潜在靶点。

表2 疾病相关的部分粪便生物标记物Table 2 Disease-related fecal biomarkers
在一定区域内,粪便微生物群落结构组成具有宿主的特异性(Erenet al.,2015)。检测环境中具宿主特异性的粪便微生物,可以追溯污染物来源,因此,此类粪便微生物可以被认定为污染溯源标记物。粪便样本中不仅含有微生物及其食用的动植物的遗传物质,还存在宿主脱落的肠道细胞。线粒体基因进化速率高、特异性较高(Brownet al.,1979),因此,粪便中宿主的mtDNA 也常作为标记物被用于动物物种鉴定及追溯污染物来源的宿主标记物,部分经典标记物如表3所示。目前的研究尚未发现任何一种标记具有绝对的宿主特异性,且一个标记物很难同时具有高敏感性和高特异性(Zhanget al.,2020)。

表3 部分粪便溯源标记物Table 3 Fecal markers for source tracking
4 宿主群体遗传评估
基于粪便样本中宿主的遗传信息,可以对研究动物进行群体遗传学方向的研究,遗传多样性、种群分化、种群遗传结构、环境异质性影响、种群动态和基因流等,进而对动物及其生境进行管理和保护(Yamazakiet al.,2011;周芸芸等,2014;Ntieet al.,2017;Baaset al.,2018)。
由于粪便含有宿主的遗传物质,基于粪便样本可以对宿主进行性别鉴定、物种鉴定、微卫星(simple sequence repeat,SSR)分型或单核苷酸多态性(single nucleotide polymorphism,SNP)分型、种群遗传多样性评估等群体遗传学研究。与性别决定相关的基因有SRY、SOX3、NR5A1和WT⁃1等,其中的SRY位于Y染色体上,是使用最为广泛的性别鉴定基因(Guneset al.,2016)。SSR 和SNP 是人们在种群评估中使用较多的2种技术,都有其自身的优势和局限性。SSR 筛选位点的费用、所需DNA 质量、操作要求和评估需要的位点数量更低,而SNP由于通量更高,更为高效(Grover&Sharma,2016)。在国内,用于评估大熊猫遗传多样性的整个SSR体系已发展得非常成熟并一直被优化(寇洁等,2022)。由于粪便中的宿主DNA 通常是碎片化的,直接从粪便中提取遗传物质往往只适用于基因片段的研究,但随着DNA 富集技术的发展,实现了从粪便样品中高效捕获宿主基因组DNA 及宿主全基因组测序数据(王李玲等,2021),可以在动物的保护基因组学等方向上进行深入研究(Fuentes-Pardo& Ruzzante,2017;Orkinet al.,2021)。王李玲等(2021)对富集粪便中宿主DNA 的方法(杂交捕获法和甲基化CpG 岛富集法)进行了深入分析和比较,甲基化CpG 岛富集法近几年才出现,稳定且成本低,富集效率与样品CpG 甲基化程度成正比。Chiou 和 Bergey(2018)利用Papio属狒狒的粪便样本对改进的甲基化CpG 岛富集法进行了验证。目前,此方法应用最多。Tyagi 等(2022)通过该方法从粪便样本中获得了丛林猫Felis chaus的全基因组SNP 数据。Taylor 等(2022)研究称,可以不进行宿主基因组DNA 捕获,直接从粪便的粘膜层中提取DNA 测序,进而获得了驯鹿Rangifer tarandus的全基因组数据。
5 总结和展望
粪便样本可提取的分子遗传信息较多,是非常便利的无损伤取样获得的样本,适用于野生动物、放牧动物、家养动物,甚至人类的研究,且各种测序技术和组学技术的发展也降低了检测成本,提高了检测效率。但是,粪便样本的使用也存在许多限制,比如粪便新鲜程度、保存时间及温度、环境污染等,都可能影响粪便中易挥发的代谢物以及核酸的降解,降低检测结果的准确率。此外,DNA 提取方法、标记位点、测序方法及深度、分析方法等不同也会导致检测结果出现较大差异(田新民,张明海,2008;单磊等,2018)。因此,尽量使用无菌器具收集新鲜粪便,是提高结果准确性的基础。研究人员已经对粪便样本保存、DNA 提取、PCR扩增等实验技术持续进行优化,比如粪便样本保存液的研发,DNA 提取方法、基因分型技术体系的优化,一代测序与二代测序结合检测,数据分析方法改进等。相信随着实验和检测方法的完善,对粪便样本遗传信息的提取将更为全面。
