缺氧诱导的线粒体自噬与糖代谢重编程对胃癌前病变影响的研究进展
2022-11-27张家祥周永学闫曙光赵唯含
张家祥,周永学,闫曙光,赵唯含,董 汾
1.陕西中医药大学基础医学院,陕西 咸阳 712046;2.陕西中医药大学附属医院消化科,陕西 咸阳 712000;3.陕西中医药大学第二附属医院肿瘤科,陕西 咸阳 712000
胃癌前病变(gastric precancerous lesions,GPL)主要表现为慢性萎缩性胃炎(chronic atrophic gastritis,CAG)伴不同程度的肠上皮化生(intestinal metaplasia,IM)与异型增生,是一种在胃癌发生前由幽门螺杆菌(helicobacter pylori,H.pylori)感染、炎症、遗传等多种因素引发的长期、多阶段,并伴随缺氧的复杂病理学过程[1]。缺氧是引起线粒体自噬及糖代谢重编程的重要条件,但GPL过程中的线粒体自噬却受到了抑制[2];与此同时,糖代谢方式也在该疾病进程中发生重编程,氧化磷酸化供能转变为糖酵解[3],上述改变最终促进了GPL异型细胞的增殖,并与胃癌的发生高度相关。线粒体自噬及糖代谢重编程作为当下研究热点,针对此二者的靶向治疗可能是有效的GPL与胃癌防治策略,本文将就其对GPL影响的研究进展进行综述。
1 GPL及其缺氧微环境的形成
根据Correa假说,胃癌沿“慢性浅表性胃炎→慢性萎缩性胃炎→IM→异型增生→胃癌”的“炎-癌链”转化[4]。GPL是该链的关键过程,虽属于良性病变,但包含了炎症、氧化应激、DNA损伤等一系列病理学改变,与胃癌具有相似的细胞异型性、快速增殖、凋亡抑制及耐药性。因此,GPL的早期诊治对防止病情进一步发展,降低胃癌发生率意义重大[5]。慢性炎症是GPL的重要特征之一,长期的炎症使大量炎症细胞、细胞因子、介质充斥于组织中,形成适合肿瘤细胞生长的炎症微环境[6]。其一方面持续激活炎症信号转导通路,直接损伤细胞、DNA,诱导致癌突变;另一方面,增强的炎症反应大幅提升细胞代谢水平,使氧耗增多,形成了低氧、低灌注的组织微环境。胃黏膜低氧状态下,血管内皮细胞生长因子(vascular endothelial growth factor,VEGF)活化,诱导功能障碍新生血管生成,加剧了组织缺氧[7-8]。值得注意的是,VEGF的表达受到了缺氧状态下激活的缺氧诱导因子1α(hypoxia inducible factor 1α,HIF-1α)的直接调控,HIF-1α是一种由826个氨基酸残基构成的相对分子质量为120×103的蛋白质[9],是重要的缺氧适应调节因子,主要参与诱导缺氧状态下的自噬与糖代谢重编程。研究[10]发现,随着大鼠GPL程度加深,胃黏膜缺氧加剧,内皮细胞增殖与微血管生成速度加快,同时HIF-1α与VEGF在胃黏膜的表达水平也逐渐升高。GPL胃黏膜持续的缺氧状态为后续自噬与糖代谢重编程提供了有利条件。
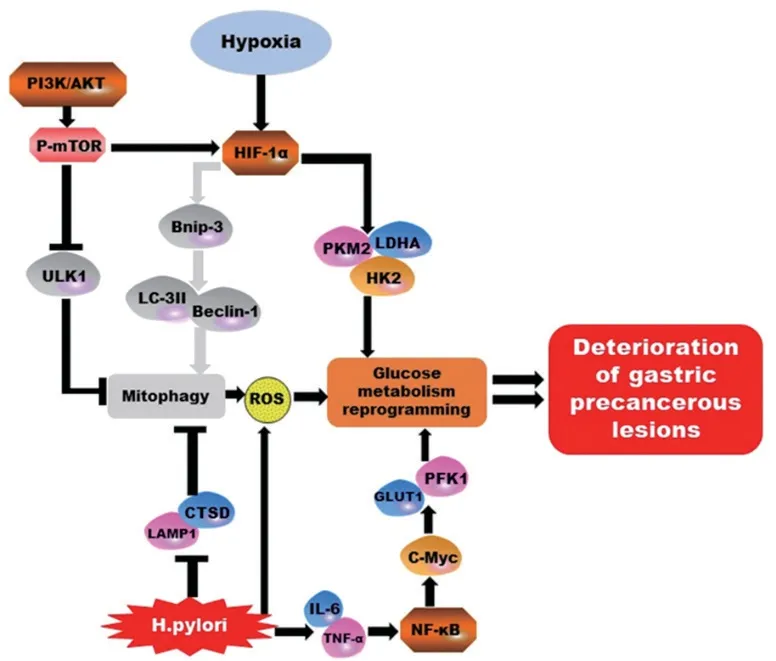
图1 自噬与糖酵解在GPL阶段作用原理模式图Fig.1 A brief schematic diagram of the interaction principle between autophagy and glycolysis during GPL
2 缺氧微环境对线粒体自噬与糖代谢重编程的诱导
GPL的微环境缺氧是引起线粒体受损或功能异常的主要原因,此时会诱导自噬启动。首先,在受损线粒体外围会形成自噬前体,逐步包绕、延伸为环状双层膜结构自噬体;之后与细胞内溶酶体结合为自噬溶酶体;最终消化受损线粒体并生成ATP再利用,这一过程被称为线粒体自噬[11-12]。缺氧状态下自噬的诱导主要通过HIF-1α完成[13],其与含有低氧反应元件(hypoxia-responsive element,HRE)的自噬关键蛋白BNIP-3结合促进其二聚化激活以诱导自噬;BNIP-3含蛋白微管相关蛋白1轻链3(microtubule-associated protein 1 light chain 3,LC-3)结合域(LC-3-interacting region,LIR),用来与LC-3结合启动自噬[14];同时自噬的发生还伴随自噬相关基因(autophagy-related genes,ATG)、Beclin-1等表达升高以及自噬底物p62表达降低。线粒体自噬的启动有利于减少损伤线粒体与活性氧(reactive oxygen species,ROS)累积,是细胞自我调控的重要机制之一。
GPL的缺氧状态还能诱导细胞糖代谢重编程的发生,即由于GPL组织氧含量降低不能满足氧化磷酸化的三羧酸循环供能,故而转向糖酵解快速且少量地生成ATP,并产生乳酸。糖酵解是葡萄糖转运体将葡萄糖从细胞外转运入细胞内,经己糖激酶2(hexokinase 2,HK2)、磷酸果糖激酶(phosphofructokinase,PFK)及丙酮酸激酶M2(pyruvate kinase M2,PKM2)等糖酵解酶分解代谢生成丙酮酸的过程[15]。在有氧条件下丙酮酸进入线粒体参与氧化磷酸化[16];而在缺氧状态下,丙酮酸则通过糖酵解途径生成乳酸。糖代谢过程改变与HIF-1α密切相关,由于HRE结合域还存在于大多数糖酵解相关酶的编码基因启动子区,所以这利于HIF-1α识别并促进参与糖酵解的HK、乳酸脱氢酶A(lactate dehydrogenase A,LDHA)、PKM2以及乳酸转运蛋白MCT1、MCT4、葡萄糖转运蛋白(glucose transporter,GLUT)等的表达[17-18]。随着糖酵解水平逐步提高,ROS与乳酸在组织中大量堆积[19],ROS稳定了HIF-1α表达,促进糖代谢重编程的持续发生[20]。
3 线粒体自噬与糖代谢重编程影响胃癌前病变的研究进展
3.1 线粒体自噬抑制促进胃癌前病变进展
正常胃黏膜细胞线粒体能通过自噬及时清理受损部分及个体,维持了线粒体正常功能与细胞稳态;此外,通过限制基因组损伤、抑制基因突变维持基因组稳定性。GPL缺氧微环境利于诱导线粒体自噬,其激活对防止炎症氧化应激增强、基因组不稳定累积及异型细胞增殖具有重要意义,提早避免了癌变危机[21]。然而实际的情况却与此相反,自噬在GPL过程中受到了抑制,GPL胃黏膜组织中自噬相关的Beclin1、LC-3ⅡmRNA或蛋白随病变的加重表达逐渐降低,并伴有自噬小体数量减少与自噬底物p62表达增多[22-23]。研究[24]发现,自噬的抑制使线粒体损伤聚集,增加了p62累积与ROS生成,它们分别通过激活NF‐kB信号转导通路与影响线粒体脱氧核糖核酸功能使ROS、肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)、白细胞介素(interleukin,IL)-6、IL-1β、IL-1等促炎因子大量释放并加剧氧化应激;这进一步造成了DNA损伤、修复障碍及基因组的不稳定,引起G:C-T:A碱基颠换,提高了致癌风险[25];同时,DNA损伤增多与错误修复会激活DNA依赖性蛋白激酶(DNA dependent protein kinase,DNA-PK)进而活化p53[26],使下游Fas、FasL、Bax等促凋亡基因启动胃黏膜细胞凋亡程序,加剧GPL胃黏膜“凋亡-增殖”失衡[27]。除此之外,自噬的抑制还造成了H.pylori及其毒力因子的持续定植与致病[28]。
GPL中自噬抑制的原因涉及诸多方面,目前尚未明确具体原因,但H.pylori感染、磷脂酰肌醇3-激酶(phosphoinositide3-kinase,PI3K)/蛋白激酶B(protein kinase,AKT)/哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)信号转导通路激活以及HIF-1α/BNIP-3信号转导通路异常等因素可能发挥了重要作用。H.pylori是胃癌的Ⅰ类致癌因子,普通人群中78%的胃癌与感染H.pylori相关[29],是正常胃黏膜病变进展至胃癌的始动力,且H.pylori毒力因子介导了自噬的抑制。毒力因子CagA与VacA能通过对溶酶体相关膜蛋白1(lysosomal associated membrane protein 1,LAMP1)、溶酶体钙离子通道1(transient receptor potential mucolipin 1,TRPML1)及组织蛋白酶D(cathepsin D,CTSD)等的负调控限制溶酶体成熟、抑制自噬关键蛋白激活,这导致自噬溶酶体消化功能异常,自噬无法正常进行[30]。近期多项研究[31-32]表明,PI3K/AKT/mTOR信号转导通路在GPL以及胃癌组织中被激活,并通过磷酸化下游的mTOR抑制自噬调控因子ULK1进而抑制自噬启动[33-34]。此外,自噬的抑制可能还涉及HIF-1α/BNIP-3信号转导通路功能失常,HIF-1α可能更倾向于激活糖酵解相关酶使BNIP-3活化减弱从而抑制自噬;这一推测尚未被证实,但主要依据是HIF-1α上游信号及胃部炎症微环境等因素促其激活下游糖酵解相关酶及诱导糖酵解的发生,这可能导致BNIP-3无法被激活。
3.2 糖代谢重编程促进胃癌前病变进展
在组织细胞的缺氧状态下,氧化磷酸化重编程为路径更短的糖酵解代谢供能,虽然ATP生成数量不及氧化磷酸化,但速率更快[35]。所以,高速率产能的特性使得肿瘤细胞中即使存在氧,也依然倾向于在细胞质中进行糖酵解供能促进其生长、增殖。这一现象被称为“Warburg效应”或“有氧糖酵解”[36]。缺氧利于糖酵解启动,正常胃黏膜细胞在H.pylori、炎症等因素作用下演变至GPL,此时胃黏膜氧化应激程度不断增强;而线粒体作为氧化应激的靶细胞器,能敏锐地感受体内氧浓度变化,若氧供应不足,则会抑制线粒体氧化磷酸化电子传递链功能与ATP生成,引起ROS产生并大量蓄积[37],导致三羧酸循环抑制[38],细胞能供不足,此时代谢方式重编程为糖酵解。如前文所述,H.pylori感染及自噬抑制在GPL胃黏膜中均能引起ROS大量分泌,这些ROS可能也通过稳定HIF-1α表达、抑制三羧酸循环从而提高糖酵解活性。
GPL的发生、发展与糖代谢紊乱关系密切[39]。目前认为,GPL的胃黏膜缺氧微环境是糖酵解激活的良好条件,而糖酵解主要是为了在缺氧状态下,满足GPL异型细胞的生长、增殖需求,以及促进形成肿瘤微环境。糖酵解的激活与负调控自噬的PI3K/AKT/mTOR信号转导通路的活化密切相关,由于该信号转导通路在GPL胃黏膜组织中被激活,所以造成GPL细胞葡萄糖摄取增加,上调HIF-1α转录与翻译水平可促进糖酵解相关基因转录[40-41],并促进了HIF-1α依赖的LDHA和PDK1过表达,减弱线粒体氧化磷酸化功能。与此同时,ROS也通过稳定HIF-1α,提高糖酵解关键酶及相关蛋白表达[32]。LDHA是GPL组织缺氧、糖酵解增强以及不良预后的标志物,作为糖酵解最后一步限速步骤调节酶,能催化丙酮酸产生乳酸[42-43]。乳酸通过MCT排出细胞外,使胃黏膜上皮细胞及DNA损伤程度加重,并在其局部创造利于GPL及肿瘤细胞增殖、侵袭的酸性环境。最新研究[44]发现,GPL胃黏膜组织中参与乳酸转运的关键载体蛋白MCT1、MCT4及其辅助因子CD147相较于正常胃黏膜表达升高,这意味着GPL胃黏膜上皮细胞可能存在较高的糖酵解水平及乳酸含量。可见GPL胃黏膜缺氧利于糖代谢重编程,并因此促进了异型细胞增殖与疾病进展,而PI3K/AKT/mTOR信号转导通路、HIF-1α、ROS以及乳酸在该过程中发挥了重要作用。
4 胃癌前病变中线粒体自噬与糖代谢重编程的相互作用
阐明GPL阶段线粒体自噬与糖代谢重编程的具体关系、作用原理可能有助于进一步探索逆转GPL的方法。最新一项关于肝癌的研究[45]表明,肝癌细胞自噬活性的增强抑制了糖酵解水平,进而使肿瘤细胞增殖速度降低。这提示我们思考线粒体自噬与糖代谢重编程是否在GPL中也具有相似关系?通过文献回顾发现,前者的逐步抑制与后者活性的持续增强,利于GPL的恶化,而这可能与此二者间的相互作用及联系相关。
4.1 线粒体自噬与糖代谢重编程在不同胃病理阶段作用的关系
正常胃黏膜进展至GPL的过程是动态可变的,线粒体自噬与糖代谢重编程在其中任何一个阶段的活性水平也不相同,可作为胃黏膜不同病理阶段的反映。具体来讲,生理状态下的胃黏膜组织较少出现缺氧,线粒体自噬并未被充分调动从而维持着基础水平,而糖代谢也以氧化磷酸化为主,所以上述二者在正常胃黏膜环境中可能并不存在相互影响的关系。但从正常胃黏膜发展至慢性浅表性胃炎及CAG的过程中,H.pylori感染等因素引起了胃黏膜炎症[46],毒力因子VacA会促进胃黏膜上皮细胞自噬水平短暂上升,之后毒力因子逐渐抑制了自噬,这是H.pylori促进胃部炎症与病理进展的重要方式之一[47];而当病变进展至CAG,若未能及时根除H.pylori,细菌则与激活的PI3K/AKT/mTOR信号转导通路一同引起自噬活性进一步抑制;与此同时,自噬抑制使ROS累积增多,促进了HIF-1α的稳定表达与糖代谢的重编程,并加剧胃黏膜炎症反应与损伤。
随着病变程度加重,IM、异型增生阶段的胃黏膜细胞异型性更加明显,并开始具备了部分与肿瘤相似的特征,如异型细胞的大量增殖、凋亡减少等。自噬相关蛋白在重度异型增生及晚期GPL胃黏膜组织中的表达也较早期显著下调,自噬小体与自噬通量均明显减少[2],这导致了基因组稳定性的破坏以及更严重的胃黏膜损伤。而持续的H.pylori感染、PI3K/AKT/mTOR信号转导通路活化均促进了GPL异形细胞糖酵解,为其细胞增殖提供了所需能量。可见从正常胃黏膜进展至重度GPL,甚至胃癌阶段,自噬水平在多种因素影响下持续降低,而糖酵解水平随病变进展程度则逐步提高,这些改变最终导致了晚期GPL组织的癌症表型。
4.2 线粒体自噬与糖代谢重编程相互作用原理
目前关于线粒体自噬与糖代谢重编程的相互作用关系及造成上述病理学进展的具体机制尚未被阐明,但越来越多的研究为二者的联系提供了依据。程雪等[48]发现GES-1细胞沉默HK2+H.pylori组LC-3Ⅱ、p62蛋白表达较阴性对照+H.pylori组分别升高与降低,自噬增强;由于HK2是糖酵解的重要限速酶,表达水平与糖酵解活性呈正相关,这意味着自噬水平的升高可能抑制了糖酵解活性。与此相反的是,一项研究[49]发现BNIP-3敲除小鼠原代成纤维细胞中线粒体自噬水平降低,功能障碍线粒体与ROS増多,并使HIF-1α表达上调,促进了糖酵解;而代谢产物质谱分析结果显示,BNIP-3缺失小鼠体内糖酵解中间体总水平较高。上述研究均表明自噬的抑制可能使糖酵解增强,反之则减弱,二者活性水平此消彼长,然而相关研究有限,还需要对GPL阶段二者的具体作用关系深入探索。
目前而言,缺氧、PI3K/AKT/mTOR信号转导通路、H.pylori、HIF-1α及ROS均参与了GPL进展并调控着自噬与糖酵解,它们可能是联系此二者的关键。缺氧在GPL胃黏膜中提供了利于自噬或糖酵解发生的环境,而H.pylori的持续感染可能充当了调控二者的“开关”,抑制了自噬并促进糖酵解。H.pylori能通过多种机制提高糖酵解活性,其抑制自噬后p62、ROS的增加及CagA、VacA等毒力因子的参与会导致GPL胃黏膜炎症反应加剧,此时炎症水平的升高引起了糖酵解的高通量[50]。近期一项研究[51]发现,毒力因子CagA能促进早期胃癌组织中PKM2的高表达,说明CagA可能早在GPL阶段就提高了细胞糖酵解水平,而这种变化可能就与CagA引起的炎症有关。TNF-α、IL-6、IL-1β等促炎因子能激活核因子κB(nuclear factor kappa-B,NF-κB)、信号转导及转录激活蛋白3(signal transducer and activator of transcription 3,STAT3)信号转导通路进而促进c-Myc与HIF-1α的共同靶基因GLUT1、HK2、PFK-1等转录,协同促进了癌前及肿瘤细胞糖酵解与乳酸生成[52]。此时PI3K/AKT/mTOR信号转导通路的活化不仅发挥了抑制自噬与诱导糖酵解启动的效应,同时也为上述炎症信号间接激活HIF-1α启动糖酵解提供了有利的路径[53]。而ROS与糖酵解、自噬水平变化间的正、负相关性说明ROS可能也影响了GPL中HIF-1α对下游信号的选择性调控。
在多种因素共同作用下,即使胃黏膜缺氧,BNIP-3可能也由于无法被活化而造成自噬诱导失败,受损的线粒体、ROS不断累积,HIF-1α在上述信号转导通路及细菌影响下,促进GPL糖酵解水平的持续升高,形成恶性循环。值得注意的是,中国作为胃癌高发病率国家,感染H.pylori在胃癌患者中极为普遍,胃黏膜细胞线粒体自噬可能在感染后的炎症初期就受到了抑制;而随着时间的延长及分期的升高,自噬抑制效应更加明显,糖酵解被彻底激活,促进了疾病恶化。
5 总结与展望
GPL病理过程的缺氧微环境引起糖代谢重编程增强,但线粒体自噬水平却减弱,这种变化能促进GPL的恶化以及胃癌的发生。作为缺氧诱导自噬的关键,HIF-1α在GPL过程中促进糖酵解的效应可能与H.pylori、PI3K/AKT/mTOR信号转导通路、线粒体自噬抑制、胃黏膜炎症微环境及ROS增加密切相关,尤其是H.pylori与PI3K/AKT/mTOR信号转导通路。近年来我们对自噬与糖酵解之间关系的认识已经取得了长足进步,但仍存在诸多问题:①并未深入研究不同程度GPL中自噬与糖酵解的关系,及二者对GPL的影响;② GPL的胃黏膜包含了正常的胃黏膜上皮细胞与GPL异型细胞,尚不明确糖酵解与自噬对上述两类细胞的具体影响;③GPL在缺氧微环境下还存在除HIF-1α外的多种信号分子活化-抑制紊乱,但目前涉及其他信号分子在GPL缺氧状态下对自噬或糖酵解影响的研究较少。
自噬与糖酵解对GPL及胃癌的影响是关键性的,今后GPL的治疗不仅要积极根除H.pylori,还应重视调控线粒体自噬水平,改善胃黏膜缺氧,防止炎症进一步加剧,抑制参与糖酵解的信号转导通路异常活化,以求全方位阻断HIF-1α对糖酵解的促进作用。目前,特异性调控自噬与糖酵解过程的药物研究日益受到重视,还应开展更多基础性的实验研究明确两种生物学过程在GPL中的具体作用关系及分子机制,使药物的研发与GPL的治疗有的放矢。
利益冲突声明:所有作者均声明不存在利益冲突。
