合理氮肥用量改善冬小麦土壤耕层细菌群落结构及理化性质研究
2022-11-25高志强杨珍平乔月静
刘 琪,高志强,杨珍平,乔月静
(山西农业大学农学院,山西太谷 030801)
0 引言
土壤微生物是地球化学循环的主要推动力,在土壤生态系统中起着至关重要的作用。微生物群落的组成以及功能对于维持土壤生态系统的稳定起着重要的作用[1-2]。研究表明,土壤微生物中占比最多的细菌几乎参与了土壤中所有的生物化学过程,在物质循环转化过程中扮演重要的角色[3]。施肥是农业生产中改善作物营养以及提高作物生产力必不可少的环节,同时也会影响土壤微生物的群落结构。黄土高原地区是北方冬麦的主要产区之一,但小麦平均单产却低于全国平均水平,为了提高产量,氮肥施用往往会过量,造成生态压力[4]。因此,研究不同施氮水平下土壤细菌群落结构以及理化性质的演替规律,对选择合适的施氮量以及改善土壤生态系统有重要意义。
土壤微生物群落的变化会影响植物的生长以及养分利用效率[5],肥料的施用会改变土壤微生物群落结构及其活力,引起土壤肥力的差异,进而影响土壤结构以及植物的生长发育[6]。适当的增加施氮量,细菌群落丰富度也会随之增加[7],还可以改变细菌门、属水平上的群落组成[8-9],显著降低了土壤真菌的相对丰度,增加了细菌的比例[10]。施肥还会改变土壤的理化性质和酶活性。与不施肥相比,适量施氮会使土壤酶活性提高[11],显著增加土壤速效氮、磷、钾及有机碳含量[12],同时影响水稳性团聚体平均重量直径[13]。
对于黄土高原地区冬小麦土壤细菌群落的研究,前人侧重于对小麦土壤细菌群落结构的生物地理分布格局或某一单一生态系统进行研究[14-15],而对不同施氮量下细菌群落结构的研究较少,在一定程度上限制了不同施氮量对土壤细菌群落结构影响的深入理解。因此,本研究以黄土高原地区小麦土壤为研究对象,结合高通量测序,分析了5种氮肥水平下土壤细菌群落结构的变化及其与理化性质的关系,以期为该地区施肥制度以及土壤生态系统的可持续提供理论依据。
1 材料与方法
1.1 试验地概况
试验在山西省运城市闻喜县后宫乡上院村(34°35'N,110°15'E)于2018—2019年进行。试验地属典型暖温带大陆性季风气候,年平均气温12℃,年平均降水量500 mm。土壤类型为石灰性褐土。试验前耕层(0~20 cm)土壤基础肥力:pH 7.93,有机质12.07 g/kg,全氮1.35g/kg,速效磷16.28mg/kg,速效钾218.76mg/kg。
1.2 试验设计
供试冬小麦品种为‘良星99’。播前浇足底墒水(750 m3/hm2),于2018年10月11日采用施肥播种一体机进行播种并一次性基施氮磷钾肥。播种方式为条播,行距为18.75 cm。设置5个施氮(N)量梯度,分别为:0 kg/hm2(N0)、90 kg/hm2(N6)、180 kg/hm2(N12)、240 kg/hm2(N16)、300 kg/hm2(N20),其中N0为对照。重复3次,共计15个小区,小区面积为2.5m×20m=50m2,完全随机区组排列。所有小区的磷、钾肥施用量均为150 kg P2O5/hm2和150 kg K2O/hm2。2019年6月7日收获。田间管理同常规大田生产。试验地前茬为玉米(Zea maysL.)。
1.3 土壤样品采集
于2019年6月用灭菌土钻采集冬小麦收获后0~20 cm土样,每个小区取3个点混合成一个样品,用恒温箱保存并运输回实验室,将样品过2 mm筛,剔除石砂和植物残渣等杂物,一部分-80℃保存,用于土壤微生物分析,另一部分土样风干后用于测定pH、全氮和土壤酶活。于收获期用环刀法取0~20 cm土样测定土壤容重和土壤重量含水率。于收获期采集0~20 cm原状土样,自然风干后剔除石砂和植物残渣等杂物,并按土壤自然裂痕将大土块剥离为1 cm3左右,用于测定土壤团聚体。
1.4 测定项目及方法
1.4.1 土壤pH、全氮和土壤酶活性 土壤pH采用电位法(水:土=2.5:1)测定;全氮用元素分析仪vario MACRO cube测定。土壤酶活性(蔗糖酶、脲酶和磷酸酶)用关松荫的方法测定[16]。
1.4.2 土壤容重和土壤重量含水率 土壤容重由105℃烘干后的土壤重量和环刀体积决定,计算公式如式(1)所示。

式(1)中BD为土壤容重(g/cm3);M2为烘干后土壤与环刀的质量(g);M0为环刀质量(g);V为环刀体积(cm3)。
土壤重量含水率利用重量法计算,计算公式如式(2)所示。
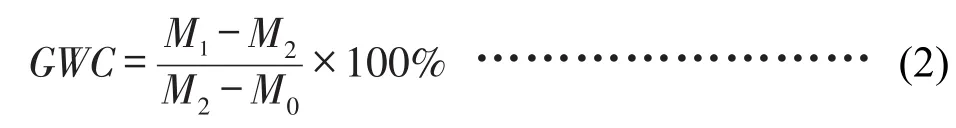
式(2)中GWC为土壤重量含水率(%);M1为鲜土与环刀的质量(g)。
1.4.3 土壤团聚体 土壤团聚体用“NYT 1121.19—2008土壤检测第19部分:土壤水稳性大团聚体组成的测定”方法测定土壤水稳性团聚体的平均重量直径[17-18]。
土壤水稳性团聚体的平均重量直径(MWD)计算公式如式(3)所示。

1.4.4 DNA提取 土壤总DNA使用Fast DNA SPIN Extraction Kits(MP Biomedicals,Santa Ana,CA,USA)试剂盒,按照试剂盒提取步骤进行。用琼脂糖凝胶电泳检测提取DNA的纯度和完整性,用NanoDrop ND-1000检测提取DNA的浓度和纯度。
1.4.5 高通量测序分析 以提取的土壤微生物总DNA为模板,利用通用引物338F(ACTCCTACGGGA GGCAGCA),806R(GGACTACHVGGGTWTCTAAT)对细菌16S rRNA V3V4区进行RCR扩增。PCR扩增体系5×reaction buffer 5 μL,5×GC buffer 5 μL,dNTP(2.5 mmol/L)2 μL,上下游引物(10 μmol/L)各 1 μL,DNA模板1 μL,Q5 DNA聚合酶0.25 μL,dd H2O 9.75 μL。PCR反应条件为:98℃ 5 min;25个循环:98℃ 30 s,52℃ 30 s,72℃ 1 min;72℃ 5 min。每个样品做3次重复,混合后利用Agencourt AMPureBeads(Beckman Coulter,Indianapolis,IN)进行纯化,使用PicoGreen ds DNA Assay Kit15(Invitrogen,Carlsbad,CA,USA)检测试剂盒进行定量。样品送由上海派森诺生物科技有限公司进行序列测序分析。
1.4.6 制备标准品及标准曲线 回收PCR产物,连接至pMD19-T载体,转化至大肠杆菌感受态中,筛选阳性克隆测序,并且提取含16S rRNA基因的质粒,用超微量核酸定量仪(BioDrop DUO)测定质粒浓度,计算16S rRNA基因拷贝数,按10倍梯度稀释,共6个梯度,用来制备16S rRNA基因标准曲线。
1.4.7 荧光定量PCR用荧光定量PCR法测定细菌16S rRNA基因丰度,反应在CFX 384Touch荧光定量PCR仪上进行。反应体系为:TB Green®Premix Ex Taq™ II 12.5 μL,上下游引物各1.0 μL,DNA模板(1~10 ng)2.0 μL,最后用超纯水(dd H2O)补至25.0 μL。反应条件为:95℃ 30 s;35个循环:95℃ 5 s,55℃ 30 s,72℃ 30 s;72℃5 min。荧光定量PCR扩增效率为93.4%,R2为0.997。
1.4.8 OTU聚类分析 对原始数据进行质量控制,运用QIIME软件识别疑问序列[19]。随后,通过QIIME软件调用USEARCH检查并剔除嵌合体序列,使用QIIME软件,调用UCLUST这一序列比对工具[20],对前述获得的序列按97%的序列相似度进行归并和OTU划分,并选取每个OTU中丰度最高的序列作为该OTU的代表序列。使用QIIME软件计算多样性指数以及获取样本在门和纲分类水平上的组成和丰度分布表。
1.5 数据处理与分析
使用Origin8.0软件绘制柱状图。用Canoco4.5软件对土壤理化性质和细菌群落结构进行冗余分析(RDA)。用R软件进行主成分分析。采用SPSS 22.0软件,利用ANOVA,对不同处理的土壤理化性质、细菌分类水平的相对丰度和α多样性指数进行单因素方差分析。
2 结果与分析
2.1 土壤理化性质
施氮量对土壤理化性质的影响如表1所示,不同施氮量土壤中除了全氮(TN)差异不显著外,其他理化指标均存在显著差异(P<0.05)。增施氮肥使土壤容重(BD)显著降低3.25%~9.74%(P<0.05);但显著增加了土壤重量含水率(GWC)和土壤水稳性团聚体平均重量直径(MWD)(P<0.05)。各指标中,N12处理表现出明显的优势。

表1 不同施氮量处理的土壤理化性质
随着施氮量的增加,蔗糖酶活性、脲酶活性和磷酸酶活性呈先升高后下降的趋势,N12处理的土壤酶活性均最高,显著高于N0处理(P<0.05),其中,脲酶活性和磷酸酶活性分别显著提高47.46%和11.49%;蔗糖酶活性在N16处理中最高,但与N12处理没有显著差异,与N0相比显著提高35.41%。
2.2 土壤细菌群落结构及多样性
2.2.1 样品序列结果及多样性指数 所有样品共测得原始序列503870条,样品含有序列28861~40061条(表2)。不同处理的Chao1指数为2335~3229,ACE指数为 2521~3316,Shannon 指数为 10.26~10.41,Simpson指数为0.99826~0.99863(表2);方差分析结果表明,施氮量会显著影响多样性指标(P<0.05),与不施氮肥相比,施氮处理中Chao1指数、ACE指数和Shannon指数分别提高9.65%~38.29%、9.13%~31.57%和0.39%~1.46%。
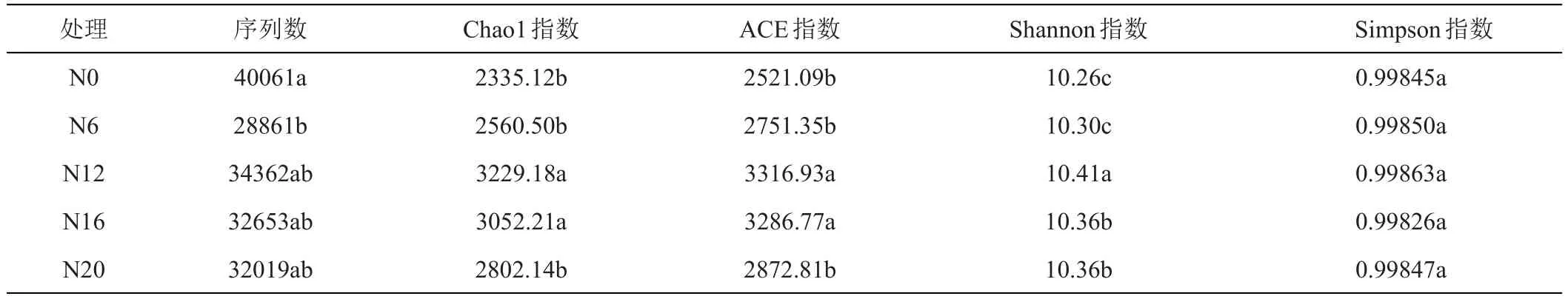
表2 不同施氮量处理的土壤细菌多样性指数
2.2.2 土壤细菌丰度 不同施氮量处理中土壤细菌16S rRNA基因拷贝数为6.47×1010~15.18×1010(图1)。随着施氮量的增加,16S rRNA基因拷贝数呈先升高后下降的趋势,在N12处理达到最大值,N0、N6和N20处理下的16S rRNA基因拷贝数没有显著差异(P<0.05)。
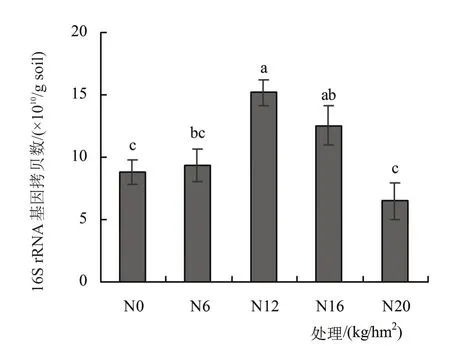
图1 不同施氮量下的16S rRNA基因拷贝数
2.2.3 土壤细菌群落组成 由图2a可知,在不同施氮量的细菌门类中变形菌门(Proteobacteria,32.20%~35.92%)、放线菌门(Actinobacteria,20.84%~27.09%)、酸杆菌门(Acidobacteria,12.70%~15.35%)、绿弯菌门(Chloroflexi,8.59% ~10.12%)和芽单胞菌门(Gemmatimonadetes,6.44%~7.77%)为土壤中优势菌门(平均相对丰度>5%),合计占的比重为84.76%~91.61%。不同施氮量间还检测到一些相对丰度较低(1%~5%)的细菌门类,如浮霉菌门(Planctomycetes,1.82% ~4.24%)、拟杆菌门 (Bacteroidetes,2.33% ~3.28%)、硝化螺旋菌门(Nitrospirae,1.23%~1.90%)、Rokubacteria (0.42%~2.48%)和匿杆菌门(Latescibacteria,0.62%~1.88%)。此外,还检测到20个相对丰度很低(平均相对丰度<1%)的细菌门类。
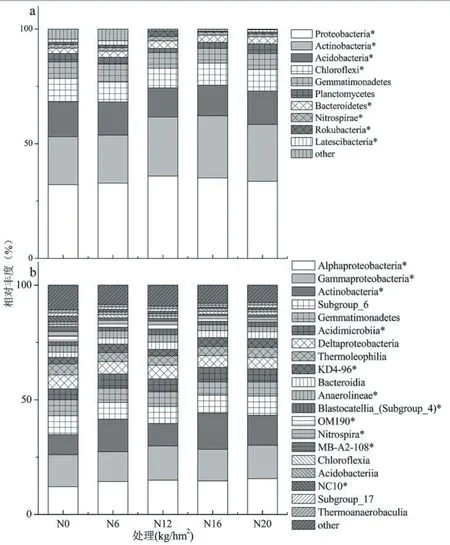
图2 不同施氮量处理的细菌门和纲水平组成
不同施氮量下变形菌门、放线菌门、酸杆菌门、绿弯菌门、拟杆菌门、硝化螺旋菌门、Rokubacteria和匿杆菌门的相对丰度有显著性差异(P<0.05),其中酸杆菌门和绿弯菌门的相对丰度在N0处理中显著高于其他处理(P<0.05),N12处理显著提高变形菌门、拟杆菌门、硝化螺旋菌门和Rokubacteria的相对丰度(P<0.05),N16处理显著提高放线菌门的相对丰度(P<0.05)。
由图2b可知,在纲水平上,得到的细菌群类中α-变形菌纲(Alphaproteobacteria,12.11%~15.65%)、γ-变形菌纲(Gammaproteobacteria,13.05%~14.94%)、放线菌 纲 (Actinobacteria,8.77% ~15.74%)、Subgroup_6(7.30% ~8.44%)、芽单胞菌纲 (Gemmatimonadetes,5.78% ~6.99%)、酸微菌纲(Acidimicrobiia,4.66% ~6.31%)、δ-变形菌纲(Deltaproteobacteria,4.93%~6.12%)为土壤中优势菌门,合计占的比重为60.85%~69.35%。不同施氮量间还检测到一些相对丰度较低的细菌类群,如嗜热油菌纲(Thermoleophilia,3.94%~4.81%)、KD4-96(2.84% ~3.82%)、拟杆菌纲(Bacteroidia,2.27% ~3.20%)、厌氧绳菌纲 (Anaero lineae,2.26% ~3.21%)、Blastocatellia(Subgroup_4,1.53%~2.29%)、OM190(0.89%~2.35%)、硝化螺旋菌纲(Nitrospira,1.23% ~1.90%)、MB-A2-108(0.61% ~2.25%)、绿弯菌纲(Chloroflexia,0.88%~1.42%)、酸杆菌纲 (Acidobacteriia,0.99% ~1.38%)、NC10(0.81% ~2.48%)、Subgroup_17(0.75%~1.43%)、Thermoanaero baculia(0.98%~1.31%)。此外,还检测到77个相对丰度很低的细菌门类。
不同施氮量下α-变形菌纲、γ-变形菌纲、放线菌纲、酸微菌纲、KD4-96、厌氧绳菌纲、Blastocatellia_(Subgroup_4)、OM190、硝化螺旋菌纲、MB-A2-108和NC10的相对丰度有显著性差异(P<0.05)。其中OM190、MB-A2-108和NC10的相对丰度在N0处理中显著高于其他处理(P<0.05),γ-变形菌纲、厌氧绳菌纲、Blastocatellia_(Subgroup_4)、硝化螺旋菌纲在N12处理中显著高于其他处理(P<0.05),放线菌纲和酸微菌纲的相对丰度在N16处理中显著高于其他处理(P<0.05),N20处理显著提高了α-变形菌纲和KD4-96的相对丰度(P<0.05)。
2.2.4 细菌群落结构分析 细菌群落的主成分分析结果表明(图3),不同施氮量处理的不同微生物群落结构有很大差异,其中,N6和N16距离较近,在同一象限,群落组成较相似;N0和N20距离较近,但是不在同一象限,且与N6、N16较远;N12处理与其他处理不同均分布于单独的象限中。

图3 细菌群落在OUT水平上的主成分分析
细菌群落结构门分类水平与土壤理化性质和土壤酶的冗余分析结果表明(图4),变形菌门、放线菌门、拟杆菌门及Rokubacteria与TN、GWC、MWD、脲酶、蔗糖酶及磷酸酶呈正相关,酸杆菌门、绿弯菌门、芽单胞菌门、浮霉菌门、硝化螺旋菌门及匿杆菌门与pH及BD呈正相关;表明土壤理化性质及土壤酶与细菌群落密切相关,其中蔗糖酶(P<0.01)和脲酶(P<0.01)显著影响了细菌群落结构(门水平)。

图4 环境因子对细菌群落结构影响的冗余分析
3 结论
与不施氮肥相比,适量施氮(180 kg/hm2)可以增加土壤重量含水率和土壤水稳性团聚体的平均重量直径,蔗糖酶、脲酶以及碱性磷酸酶活性也会升高;而过量施氮,土壤pH、容重会显著下降,土壤酶活性也会降低。增施氮肥会显著改变细菌门和纲水平上的相对丰度。
在黄土高原地区适量施氮(180 kg/hm2),可以改善土壤结构,提高土壤细菌群落多样性,为维持生态系统平衡奠定基础。
4 讨论
4.1 施氮量对土壤理化性质的影响
研究表明长期施氮使土壤pH下降[21-22],在本研究中pH没有显著变化,且pH与土壤细菌群落没有显著相关性,可能与施氮的时间长短有关,也可能是由于研究条件不同引起的,尤其是试验地土壤pH不同。研究表明,碱性土壤明显减缓了土壤pH的变化,使得土壤pH变化很小,且在施肥处理中,碱性土壤的化学性状与细菌群落结构没有明显的关系;但是在酸性和近中性土壤中,土壤化学性状与细菌群落结构有显著的相关性[23],而前人研究的土壤pH通常是酸性和近中性的[24-26]。水稳性团聚体的MWD表示了土壤的团聚度和稳定性[17],本研究表明过量施氮会破环土壤结构,导致MWD下降,这与邢旭明等[13]研究结果相似。研究表明在施氮量超过N12处理时土壤容重没有显著变化,而GWC有所下降,这可能是高氮投入导致土壤环境恶化,这与李荣等[27]的研究结果一致。
增施氮肥对土壤酶活性有显著影响,适量施氮会提高土壤酶活性。蔗糖酶、脲酶和磷酸酶在施氮量(N)超过180 kg/hm2时活性会下降,这与CHEN研究结果相似[28]。但MARKLEIN等[29]研究发现磷酸酶活性会随增施氮肥而升高,因为磷酸酶的产生需要高氮投入;在MARKLEIN的研究中虽然土壤磷酸酶平均活性升高,但是在他85个试验地中有26个的土壤磷酸酶活性降低,这可能与施肥时间、土壤肥力、微生物群落等有关。本研究发现,土壤酶活性与细菌群落相关性最高,可能是因为土壤微生物是产生土壤酶的主要原因,而且土壤酶活性可以反应土壤退化潜力[30]。
4.2 施氮量对细菌群落结构的影响
有研究表明,增施氮肥会降低土壤细菌多样性[31]。本研究中增施氮肥,细菌多样性指数呈先增加后减小的趋势,这与之前的报道一致[32]。有研究表明,氮肥添加引起的酸化作用是导致土壤微生物多样性下降的主要因素[33];也有研究表明,氮累积会刺激嗜氮微生物的增加和其他微生物的竞争,从而导致微生物多样性下降[34]。微生物多样性降低会导致陆地生态系统改变[35],所以过量施氮可能会导致生态系统不稳定[36]。
结果表明,不同施氮量处理的门和纲类群相似,土壤细菌群落对不同施氮量的反应不同,土壤细菌在门和纲水平上有显著变化(P<0.05)。在高氮处理下酸杆菌门的丰度有所下降,酸杆菌门在生态学上归类为寡营养菌门[37];在高氮处理下富营养菌门中放线菌门丰度最高,RAMIREZ等[9]观察到在施用氮肥的土壤中微生物呼吸速率较低,假设施用氮肥通过改变细菌代谢来抑制土壤微生物活性,那相对于寡营养细菌,能够降解不稳定化合物的细菌群落一定会有所增加。有研究表明,变形杆菌和酸杆菌之间的比例可表示土壤的营养状况,在贫瘠土壤和有机输入量较低的农业土壤中该比例较低,而在有机输入量较高的农业土壤中该比例较高[38-39]。研究发现N12处理的细菌群落在门水平的变形菌门显著高于其他处理,其酸杆菌门较低,所以N12处理的土壤营养状况较好。此外,本研究发现在N12处理中能够将亚硝酸氧化为硝酸盐的硝化螺旋菌属丰度最高,说明适量氮肥有助于提高土壤氮肥力[40]。在N12处理中未分类的细菌门Rokubacteria丰度显著高于其他处理(P<0.05)。有研究表明,Rokubacteria与硝化螺旋菌门密切相关,与硝化螺旋菌门不同的是,Rokubacteria基因组很大,其中GC含量高以及具有多种混合营养代谢潜力[41]。综上所述,不同施氮量可以改变土壤细菌群落结构,适量施氮会提高土壤细菌多样性。冗余结果也表明,环境因子与优势菌门变形菌门、拟杆菌门、放线菌门和硝化螺旋菌门相关性较强,这说明适量施肥对土壤细菌群落结构有良好的影响。
不同施氮量对土壤细菌群落及理化性质也有一定影响,本研究只反映了土壤细菌群落变化,未分析不同施氮量下其他种类微生物的变化,不同施氮量引起的细菌群落差异是否对其他种类微生物产生影响还有待研究。
