胱氨酸基荧光碳点的制备及其黄体酮的荧光恢复响应
2022-11-24钟榕榕翁文婷高平章柴梓欣
钟榕榕, 翁文婷*, 高平章, 柴梓欣
(泉州师范学院化工与材料学院,福建泉州 362000)
黄体酮(Progestin)主要成分为孕甾-4-烯-3,20-二酮,中国药典中对此药物的分析使用高效液相色谱法[1],也有研究采用电化学发光法[2]和示差脉冲伏安法[3],但结合灵敏的荧光光谱分析方法的研究还罕见报道。碳点(Carbon Dots,CDs)有良好的光电性能,同时具有低细胞毒性、高生物相容性等优点,已广泛应用在光电子器件、生物检测和医药分析等领域[4]。碳点的结构和性能均与所选择的碳源类型相关[5,6],而碳源和钝化剂的成分组成更是直接影响到碳点的表面基团结构。水热反应过程中引入分子结构含有氨基和巯基的碳源,可以简便快速制得氮硫掺杂的碳点,明显提高碳点的荧光量子产率[7 - 9]。Dong等[10]最早报道以半胱氨酸和柠檬酸为混合碳源,引入氮硫原子掺杂,可制备荧光量子产率高达73%的碳点。随后,此类碳点被广泛应用于痕量离子和药物的高灵敏检测,深入研究表明掺杂和柠檬酸协同作用形成的碳核型结构是高荧光量子产率根本原因[11 - 13]。
本文以胱氨酸(Cys)和柠檬酸(CA)为碳源,用一步水热法制得发光性能良好的氮硫掺杂荧光碳点CA-Cys-CDs。CA-Cys-CDs的荧光信号对Pb2+具有最敏感性响应,其荧光猝灭程度与Pb2+浓度呈现良好的线性关系。当在该体系中加入黄体酮时,导致Pb2+与碳点之间的结合能力降低,荧光信号得以恢复,基于此机理可实现对黄体酮含量的测定。
1 实验部分
1.1 主要仪器与试剂
CaryEclipse荧光分光光度计(美国,安捷伦公司);Libra紫外-可见分光光度计(英国,柏诺);FS920荧光光谱仪(英国,Edinburgh Instruments);TECNAI G2 Spirit TWIN场发射透射电子显微镜(美国,FEI公司);CHI660D电化学工作站(上海辰华仪器有限公司);Zeta电位及粒径分析仪(美国,布鲁克海文仪器公司)。
胱氨酸(Cys)、柠檬酸(CA)购自上海阿拉丁试剂有限公司;光谱纯石墨棒(>99%,北京电碳厂);Pb2+标准溶液(1 000 μg/mL)购自国家钢铁材料测试中心钢铁研究总院;黄体酮(C21H30O2,>98.0%)购自梯希爱化成工业发展(上海)有限公司;黄体酮注射液(上海金宇生物科技动物药业有限公司,规格为50 mg/1mL);AgNO3、FeCl3·6H2O、ZnCl2、AlCl3·6H2O、CuCl2·2H2O均购自上海阿拉丁试剂有限公司;其余试剂均为分析纯;实验所用水为超纯水。
1.2 实验方法
1.2.1 溶液的配制准确称取Cys 0.3025 g,用适量的NaOH溶液溶解,再用水定容,配制成pH=9.0的0.01 mol/L溶液,通氮除氧5 min,作为反应液A。另取0.0603 g CA,溶解在10 mL的水中,作为溶液B。准确称取黄体酮固体用乙醇溶解,配制成1.02×10-4mol/L的母液,现配现用。
1.2.2 电解刻蚀法制备荧光碳点截取15 cm石墨棒作为工作电极,将表面打磨至光滑,浸入电解液中10 cm,以1 cm×1 cm的铂片电极作为对电极,饱和甘汞电极(SCE)作为参比电极。三电极体系在反应液A中,在电化学工作站上采用恒定电位法电解。设定采样间隔为0.1 s,静置时间为2 s,电位6 V,电解时间3 600 s。电解过程中溶液由无色渐变为微黄色的碳点溶液,去除原料后,得到的溶液保存于4 ℃冰箱中。制备的碳点记为GQDs。
1.2.3 水热法制备荧光碳点准确移取15 mL反应液A至25 mL的不锈钢高压反应釜的聚四氟乙烯内衬杯中,通氮气除氧密封后,于210 ℃的烘箱中反应5 h,制备的碳点记为Cys-BCDs。将15 mL反应液A密封于圆底烧瓶中,于90 ℃条件下恒温水浴加热22 h,制备的碳点记为Cys-GCDs。将溶液A和B按等比例体积混合,超声混合均匀后,转移至25 mL的不锈钢高压反应釜的聚四氟乙烯内衬杯中,通氮气除氧密封后,于210 ℃的烘箱中反应5 h,制备的碳点记为CA-Cys-CDs。反应结束后冷却至室温,将反应后溶液过滤,转移至500 Da透析袋中透析2 d,得到的溶液保存于4 ℃冰箱中。
1.2.4 碳点的表征将碳点溶液在荧光光谱仪上采用不同激发波长进行荧光光谱扫描,光电倍增管电压为400 V,激发/发射狭缝为5.0 nm/5.0 nm,扫描速度为1 200 nm/min。在FLS920型稳态/瞬态荧光光谱仪测试荧光寿命曲线,采用EPLED半导体激光器为激光光源,使用双指数函数对荧光寿命的曲线进行拟合[14]。将纯化后的碳点溶液根据文献方法[15],以含硫酸奎宁的0.5 mol/L硫酸溶液作为参比物质,采用相对比较法测定碳点溶液的荧光量子产率。取纯化处理后碳点溶液用透射电镜观察碳点形貌,设置加速电压为40~120 kV,放大倍率为10~50万倍。所得图像使用Nano measure软件分析处理。取透析过碳点溶液采用Zeta电位仪表征团簇表面电荷状态和水合粒径,采用He-Ne激光器激发。
1.2.5 荧光碳点探针对黄体酮的响应分别取CA-Cys-CDs溶液1 mL置于10支10 mL的比色管,加入3 mL pH=5.72的B-R缓冲液和1 mL 5.0×10-5mol/L Pb2+溶液,用水定容到10 mL,摇匀静置后测定加入药物前后溶液的荧光光谱。同等条件下进行乙醇空白溶剂的对照试验。
1.2.6 黄体酮注射液的含量分析取黄体酮注射液10支,混合后,精密吸取药物(约相当于黄体酮50 mg)于10 mL比色管中,用乙醚溶解稀释至刻度,摇匀,在温水浴中使乙醚挥散,用乙醇振摇提取4次(第1~3次每次5 mL,第4次3 mL) ,合并提取液作为供试品溶液,按“1.2.5”方法,测定注射液样品的结果并与标示值比较。
2 结果与讨论
2.1 不同方法制备胱氨酸基碳点的光学性质
不同碳点溶液的荧光光谱和荧光衰减曲线如图1所示,它们的发光性能和结构表征数据见表1。电化学刻蚀法制备的碳点GQDs溶液的Zeta电位值达到-27.9 mV,说明碳棒上剥离下来碳纳米粒子表面呈负电荷状态。通过透射电镜表征其形貌,结果如图2(a)所示,其平均粒径约为2.60 nm,碳核具有石墨烯晶格结构。碱性胱氨酸溶液在90 ℃恒温条件下反应形成的碳点溶液Cys-GCDs呈现绿色荧光,其最佳荧光发射峰位置为508 nm。纳米分子团簇粒径约为1.81 nm,表面呈现更稳定的负电荷状态[16]。180 ℃高温下制备的碳点Cys-BCDs最佳发射峰位置为450 nm,平均粒径增大至3.32 nm,拟合平均荧光寿命值较小,这可能是高温碳化形成内核,表面基团改变导致。如果在碳源溶液中加入柠檬酸,制备的CA-Cys-CDs具有较好的稳定性,荧光强度增强近10倍。荧光量子产率高达61.7%,而且呈现一级长荧光寿命特征。说明柠檬酸分子结构有利于形成分子态碳核发光中心,明显增强荧光发射信号,与文献中描述一致[12]。

图1 不同制备方法合成CDs溶液的荧光光谱(a)和荧光衰减曲线(b)Fig.1 Fluorescence spectra (a) and decay curve (b) of CDs solutions prepared with different methodsInsert:the image of CDs solutions under ultraviolet light(from left to right:GQDs,Cys-BCDs,Cys-GCDs,CA-Cys-CDs)
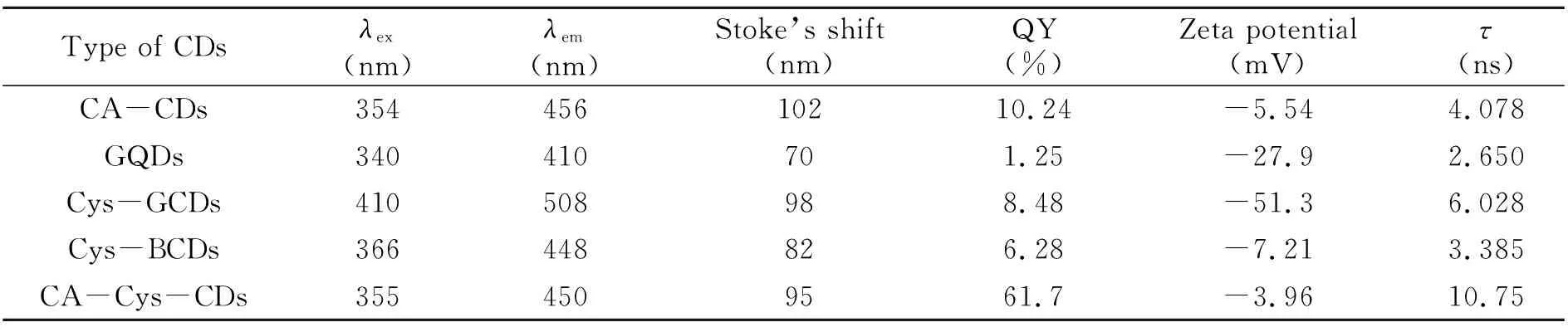
表1 不同制备方法合成CDs的荧光性质和表面电荷状态Table 1 Optical properties and Zeta potential of CDs prepared with different methods

图2 不同制备方法合成的碳点GQDs(a)、Cys-GCDs(b)、Cys-BCDs(c)和CA-Cys-CDs(d)的透射电镜(TEM)图(内插图为所对应的粒度分布直方图)Fig.2 TEM images of GQDs(a),Cys-GCDs(b),Cys-BCDs(c) and CA-Cys-CDs(d)(Inset:the corresponding particles size distribution histograms)
2.2 CA-Cys-CDs制备条件的优化
基于“2.1”的实验结果,以荧光光谱为观测手段,采用单因素法探究CA-Cys-CDs体系的合成条件。Cys碳源和CA之间的用量比例实验结果如图3(a)和3(b)所示,固定CA用量,当CA与Cys质量比为1∶5时,制得碳点溶液的荧光强度最强,表明引入Cys的氮硫原子掺杂明显提高碳点的发光性能。当Cys质量超过0.3 g之后,碳点溶液的荧光强度反而下降。这可能是因为此时体系中CA含量比例较小,在碳点形成过程中辅助作用减小导致的。若固定Cys用量,随着CA用量的增加,直接影响反应液的酸度,当pH大于5以上发射峰位置稳定在450 nm,pH小于4时碳点溶液荧光发射峰逐渐发生蓝移至425 nm。但碳点溶液不稳定,易生成沉淀。因此选择最佳条件为反应前驱体溶液的配比为1∶5,调节pH为5,温度为180 ℃,反应时间为5 h。

图3 不同原料质量比(a)、反应液pH(b),反应温度(c)和反应时间(d)的CA-Cys-CDs荧光光谱Fig.3 Fluorescence spectra of CA-Cys-CDs solution affected by dosage ratio(a),pH(b),reaction temperature(c) and reaction time(d)
2.3 CA-Cys-CDs结构的表征
采用X射线光电子能谱(XPS)对CA-Cys-CDs的结构组成进行表征,结果如图4所示。C 1s、N 1s和O 1s的特征峰分别出现在283.98 eV、399.98 eV和529.98 eV处,同时在164.85 eV处出现S 1p的特征峰。说明Cys的氨基和巯基在碳点合成过程中嵌入碳点表面形成N、S掺杂型的CDs。将纯化后的碳点溶液滴加在铜网上,并用红外灯将其烘干,对其进行了透射电镜(TEM)表征。从图2(d)中可以看出,碳点呈现大小均匀且分散性良好的圆球状分布。粒径大小在0.85~4.25 nm范围内呈现正态分布,平均粒径约为2.45 nm。
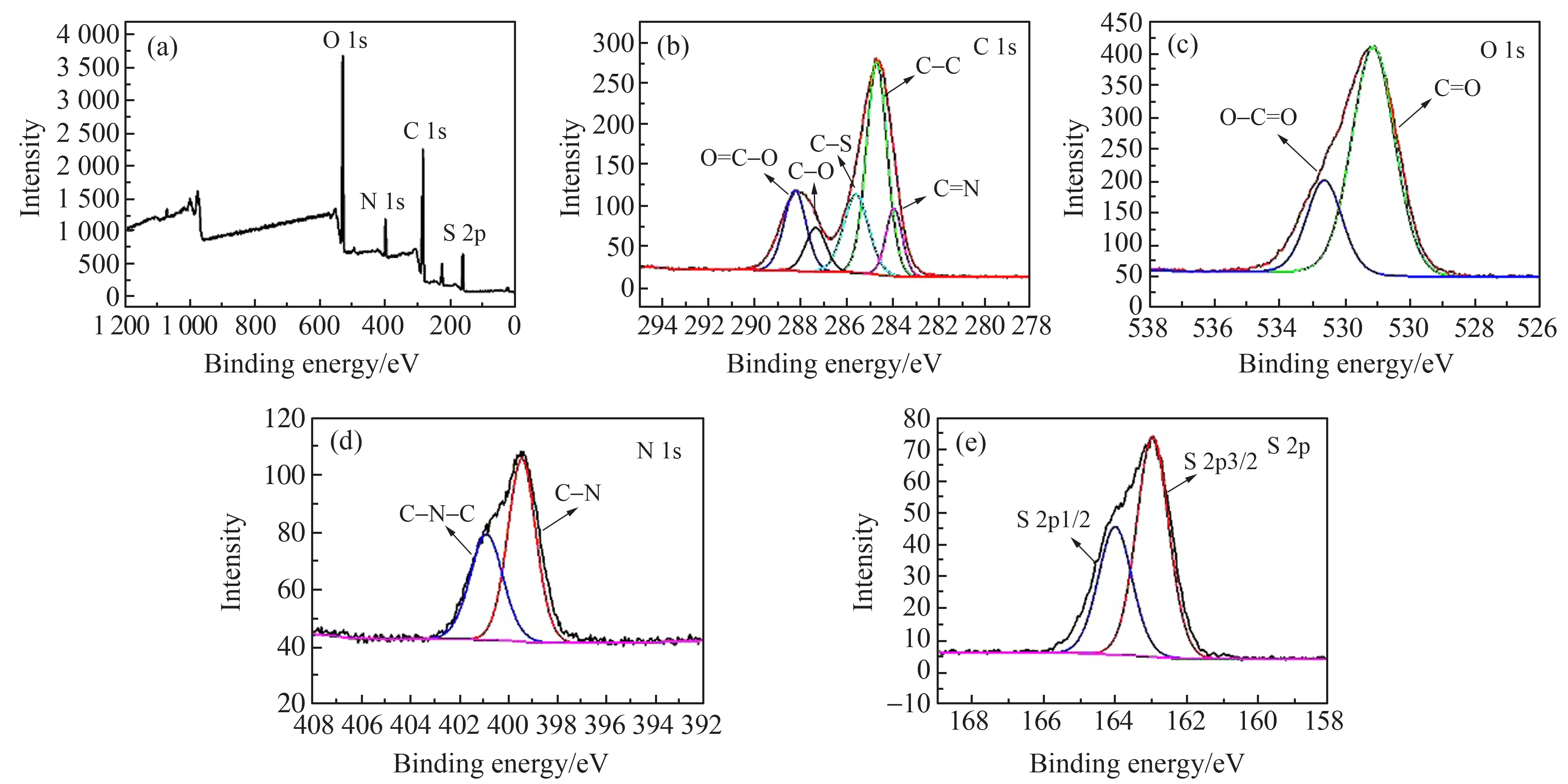
图4 CA-Cys-CDs的XPS全谱(a)、C 1s谱(b)、O 1s谱(c)、N 1s谱(d)和S 2p(e)Fig.4 XPS spectra and high-resolution XPS spectra of CA-Cys-CDs (a),C 1s (b),O 1s (c),N 1s (d) and S 2p (e)
2.4 CA-Cys-CDs碳点的光谱性能表征
将碳点溶液放在365 nm紫外灯下照射,结果表明合成的碳点溶液几乎没有光漂白性。同时考察在0.01~0.10 mol/L NaCl溶液中,碳点溶液对盐浓度的敏感程度,结果表明该碳点的荧光强度基本保持稳定,显示出优异的抗盐能力。
用不同波长的光源对碳点溶液进行荧光发射光谱的扫描。实验结果如图5所示,在280~400 nm范围内,随着激发波长的增加,荧光强度先增强后减弱,在其激发波长为360 nm时,荧光强度最大。最佳荧光发射波长在激发波长400 nm之后发生红移,表明该碳点具有分波段范围内的波长不依赖性。

图5 不同激发波长下的CA-Cys-CDs的荧光发射光谱Fig.5 Emission spectra of CA-Cys-CDs solution at different excitation wavelengths
将合成的碳点溶液冷冻干燥为固体后,考察其在不同溶剂中的溶解度。结果表明该碳点易溶于水,能溶于极性溶剂,但是不溶于非极性溶剂。进一步考察碳点在不同pH值的B-R缓冲液中的荧光光谱,结果如图6所示,荧光强度随着pH的增加先增后降,碳点溶液的荧光发射峰位置在pH=4~12范围内都稳定在450 nm,但是在较酸性条件下荧光强度急剧下降,这可能是因为碳点表面的发光基团在此pH环境中电离状态不同导致。考虑到后续碳点溶液的应用研究,所以本实验选择pH=5.72为最佳溶液环境。

图6 不同pH溶液中的CA-Cys-CDs的荧光发射光谱Fig.6 Emission spectra of CA-Cys-CDs in different pH solution
2.5 应用研究
2.5.1 CA-Cys-CDs对金属离子的响应按照实验方法,研究常见的金属离子对碳点荧光信号的响应情况,结果如图7所示。Al3+和Zn2+没有响应信号的变化,能与N原子形成配位作用的Cu2+和Ag+的响应斜率一致。该碳点表面可能存在大量的羧基和羟基,易与Fe3+结合成配合物,所以对Fe3+有一定的荧光信号猝灭效应。由于碳点结构中存在的巯基易于Pb2+形成共价键作用,导致Pb2+猝灭碳点溶液的荧光信号现象最为明显。随着Pb2+的浓度增加,碳点溶液的荧光强度逐渐下降。
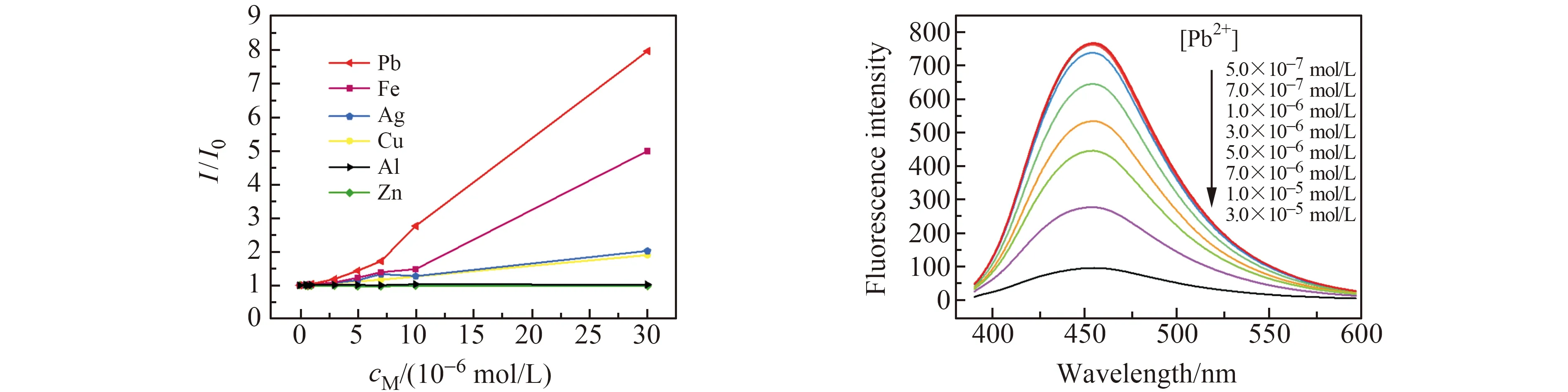
图7 CA-Cys-CDs对不同金属离子的响应趋势(a)及对Pb2+的荧光光谱(b)Fig.7 Response trend of CA-Cys-CDs to different metal ions (a) and fluorescence spectra of CA-Cys-CDs to Pb2+(b)
2.5.2 基于荧光恢复测定黄体酮按实验方法,在pH=5.72的B-R缓冲溶液中,加入碳点和Pb2+,等荧光信号稳定。增加不同浓度的黄体酮标准溶液,充分摇匀,静置1 min后,于荧光分光光度计上测定荧光强度,观察体系的荧光信号恢复程度。结果如图8所示,在9.90×10-7~1.07×10-5mol/L浓度范围内,CA-Cys-CDs-Pb2+体系荧光强度的增加程度与加入的黄体酮浓度呈现良好线性关系。线性方程为:I/I0=5.83×104c+1.02,相关系数为0.9908,方法检出限为3.7×10-7mol/L。以乙醇溶剂代替黄体酮标准溶液进行对照试验,结果表明碳点体系的荧光恢复并非溶剂效应所导致。
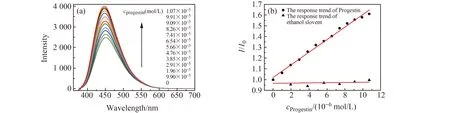
图8 CA-Cys-CDs-Pb2 +体系随着不同浓度黄体酮标准溶液的荧光光谱(a)和线性曲线(b)Fig.8 Fluorescence spectra (a) and linear curve (b) of CA-Cys-CDs-Pb2 + with different concentration of progestin
为了说明黄体酮在荧光体系中的作用机理,通过测定加入黄体酮前后溶液体系的荧光衰减曲线,拟合后数据见表2。荧光碳点体系均具有明显的二级寿命的荧光特性,当荧光碳点表面基团结合Pb2+后,发射过程可能存在一定的非辐射衰变过程,导致荧光发射信号降低,荧光寿命减少。当CA-Cys-CDs-Pb2+体系中引入黄体酮,使体系的荧光得以恢复,加入黄体酮前后体系的衰减曲线与双指数方程能够很好地契合,平均荧光寿命几乎不变,表明碳点结构状态没有明显变化。荧光信号恢复可能是由于黄体酮在碳点表面形成一定的空间位阻效应,阻碍Pb2+在碳点表面的富集。
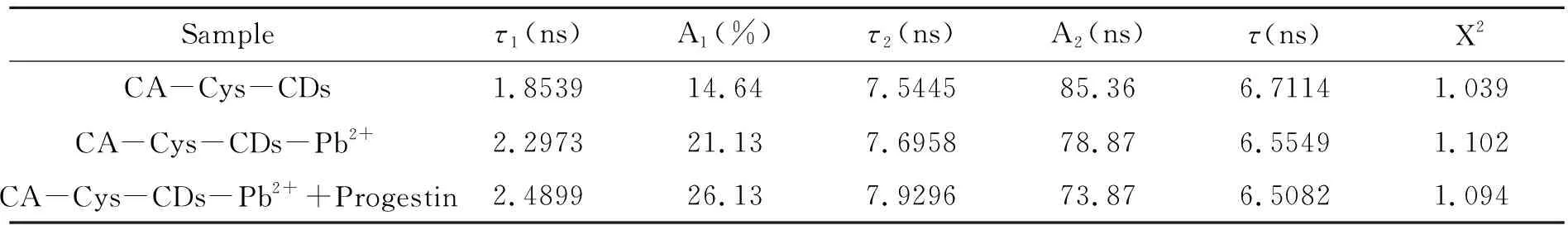
表2 CA-Cys-CDs和加入黄体酮前后CA-Cys-CDs-Pb2+溶液的荧光寿命拟合数据Table 2 Fitting value of fluorescence decay curve of CA-Cys-CDs,CA-Cys-CDs-Pb2+ with and without progestin

2.6 黄体酮注射液样品的测定
将黄体酮注射液按实验方法进行处理,准确移取供试品溶液用乙醇稀释至1.0 μg/mL,按“1.2.6”进行含量测定,按其回归方程计算黄体酮的浓度,换算药品的百分标示量为93.2%~112%,表明基于碳点的荧光信号恢复的检测方法可用于黄体酮定量的检测。
3 结论
本研究以胱氨酸为主要碳源,通过添加柠檬酸,水热反应形成氮硫掺杂的结构,明显提高了碳点的荧光性能。CA-Cys-CDs表面呈负电荷性质,最佳激发/发射波长为355/450 nm。双指数拟合后平均荧光寿命为10.75 ns,荧光量子产率为61.7%,其水溶液具有稳定的抗光漂白性和抗盐性能。在pH=5.72的B-R缓冲溶液条件下,该碳点的荧光信号可以被Pb2+所猝灭。在含有5.0×10-6mol/L的Pb2+的CA-Cys-CDs-Pb2+体系中,加入黄体酮后,因为Pb2+与碳点之间的结合能力下降,可导致体系的荧光信号得以恢复。黄体酮浓度与体系的荧光强度恢复程度呈现良好的线性关系,检出限为3.7×10-7mol/L。该实验结果可为碳点荧光探针用于药物分析提供一定的理论参考。
