CH3ONO 与OH 自由基的反应机理及动力学研究
2022-11-24赵晓霞刘子忠赵瑞生许天孜
赵晓霞,刘子忠,3,赵瑞生,许天孜
(1.内蒙古师范大学 化学与环境科学学院,内蒙古 呼和浩特 010022;2.内蒙古自治区绿色催化重点实验室,内蒙古 呼和浩特 010022;3.内蒙古自治区应用数学中心,内蒙古 呼和浩特 010022)
亚硝酸甲酯(CH3ONO)是一种常用于化学工业的反应试剂[1],同时也是平流层和对流层中的重要物质[2]。然而,CH3ONO 具有剧毒性,可引起人类呼吸的敏感性[3]。随着甲醇汽油燃料的推广使用,大量的CH3ONO 从汽车尾气排放到大气中[4-5]。这些气体进入大气后迅速光解,其产物是光化学烟雾的主要来源[6]。对于亚硝酸甲酯的光解污染已经得到广泛重视[7-8],而CH3ONO 与一些自由基的反应产物也与大气污染密切相关[9-11]。对流层中CH3ONO 的非光解主要反应路径是与OH 自由基反应,其产物CH2O 是对流层VOC 光氧化过程中关键的中间体[12],HONO 和NO 是夜间无机硝酸盐的重要前体物[13],而CH3O 自由基易在大气中氧化,影响O3和其他大气氧化剂的生成[14],因此了解CH3ONO 与OH 自由基的反应具有重要的研究价值,目前研究者发现该反应存在两种反应路径:

在过去四十多年中,有关OH 自由基与CH3ONO 的反应动力学已有大量的实验室研究。Campbell等[15]采用相对速率法,以H2O2和NO2在黑暗中非均相反应作为OH自由基的来源,在292K 下测得反应速率常数为1.51×10-12cm3·molecule-1·s-1。而Tuazon等[16]却利用N2H4与O3发生均相反应作为OH自由基的来源,得到的速率常数为1.8×10-13cm3·molecule-1·s-(1(300±3)K),几乎比Campbell报告的速率常数低10 倍。Nielsen 等[17]研究了Campbell 和Tuazon 速率常数可能存在的误差来源,但是未能找到原因。他们通过脉冲辐射-紫外动力学光谱的绝对技术和相对速率技术测得CH3ONO+OH的反应速率常数为(3.0±1.0)×10-13cm3· molecule-1· s-1,与Tuazon等的研究结果相近。Cox等[6]提出OH自由基与CH3ONO 的反应涉及氢提取和OH 自由基加成两种反应途径,根据光解反应初始阶段的NO 时间分布,提出了反应速率常数比为k1a/k1b=1.25,并认为提取和加成同样重要,但并未检测到HONO 的形成。Djegiche等[10]在CH3ONO+OH 自由基反应中首次检测到HONO,但是认为OH 加成产生的HONO 最多占CH3ONO 消耗的5%。这些实验研究为CH3ONO+OH 自由基的反应机制及动力学提供了重要信息,但是对于氢提取和OH 自由基加成途径的占比还存在不一致的看法。此外,虽然很多实验测定了反应速率,但理论上对反应机理的研究相对较少。基于此,本文采用密度泛函理论研究了CH3ONO 与OH 自由基的反应机理,并计算了各通道的反应速率常数,希望通过本研究能理解氢提取和OH 自由基加成路径的真实占比以及提供详细的反应机理,为今后的研究提供理论指导。
1 计算方法
所有计算均在Gaussian 09 程序中完成。反应势能面上所有稳定点的优化,包括反应物、反应物复合物、过渡态、产物复合物和产物都在M06-2X/6-311+G(2df,2p)水平上完成。利用振动频率以及内禀反应坐标(IRC)分析反应中的过渡态,以检验反应物复合物确实通过过渡态连结到相应的产物复合物。为了获得最佳反应路径上各稳定点更精确的电子能量,本文在优化的几何结构上采用CCSD(T)/aug-cc-pVTZ 方法进行了单点能计算。
为了评估氢提取和OH 自由基加成的占比,本文采用过渡态理论和稳态近似法计算了240~425 K 范围内反应的速率常数,并进行了隧穿校正。这种计算方法已在很多研究中报道过,预测的速率常数与实验值吻合较好。此外,为了确保结果的准确性,本文利用KiSThelP 软件对最优反应路径进行了速率计算。CH3ONO 与OH 自由基的反应以反应物复合物的形成开始,然后经过相应的过渡态生成产物,反应为:

其中,k1和k-1分别是CH3ONO 和OH 自由基碰撞的速率常数以及反应物复合物分解的速率常数,k2是反应物复合物分解为产物的速率常数。假设中间体与反应物达到平衡状态且处于稳态,则总速率常数可以表示为

根据Singleton 等[18]和Alvarez-Idaboy 等[19]的研究,由于k-1中的熵变比产物形成中的熵变大得多。因此,k-1比k2大得多,所以反应速率常数k可以表示为

Keq是平衡常数,计算公式为

其中,ΔG是反应物形成反应络合物的吉布斯自由能,kB是玻耳兹曼常数,R是摩尔气体常数,T是绝对温度,p0是大气压力。k2是反应物复合物分解为产物的速率常数,利用过渡态理论计算k2的公式为

ΔG+是反应络合物形成过渡态的吉布斯自由能,Γ是量子力学隧道效应校正

h是普朗克常数,v+是过渡态的虚频(cm-1)。
2 结果与讨论
根据C-O 键相对于N=O 键的位置的不同,CH3ONO 有顺式(cis-)和反式(trans-)两种构型,两种构象之间可以快速转换。图1 显示了cis-CH3ONO 和trans-CH3ONO 优化后的几何参数,与现有的实验结果[20]有很好的一致性。图1 中所标注所有键的键长与实验值差异小于0.003 nm,所标注键角(∠C-O-N和∠O-N-O)与实验值差异小于1.3°。计算得到trans-CH3ONO 的能量要比cis-CH3ONO 构型稳定3.56 kJ/mol,与文献[9]报道的1.59 kJ/mol 相近。这表明它们在大气中的相对丰度几乎相等。由于OH 自由基进攻CH3ONO 时既可以发生氢提取反应,又可以发生OH 加成反应,所以cis-CH3ONO 和trans-CH3ON 存在2 个不同的位点供OH 自由基进行进攻,它们分别是:OH 自由基进攻C 上的氢,即直接H 提取反应;OH 自由基进攻N 原子,即加成-消去反应。

图1 cis-CH3ONO 和trans-CH3ONO 优化后的几何构型Fig.1 Optimized structures of cis-CH3ONO and trans-CH3ONO
2.1 cis-CH3ONO 与OH 自由基的反应势能面
cis-CH3ONO 与OH 自由基反应时共存在四条反应路径,路径1 和路径2 反应机制为直接氢提取,路径3和路径4 为OH 自由基加成。图2 和图3 分别显示氢提取和OH 自由基加成路径中所有中间体优化后的分子构型及其结构参数,能量参数以及过渡态的虚频大小见表1,反应势垒图如图4 所示。

表1 cis-CH3ONO+OH 自由基反应的能量参数及过渡态的虚频Tab.1 Energy parameters and virtual frequency of transition state of cis-CH3ONO+OH radical reaction
2.1.1 氢提取 图2 显示了cis-CH3ONO+OH 自由基反应中发生氢提取机制各中间体的稳定构型,反应中的每个过渡态都有且仅有一个虚频。路径1 是OH 自由基进攻cis-CH3ONO 甲基上位于对称面两侧的氢,而路径2 是OH 自由基进攻cis-CH3ONO 甲基上位于对称面上的氢,分别形成了反应物复合物CR1 和CR2。通过对势能面分析,CR1 的相对能量为6.61 kJ/mol,比CR2 稳定2.97 kJ/mol。路径1 中,随着两个反应物分子的相互靠近,OH 自由基的O 原子提取cis-CH3ONO 的H3 原子,CR1 越过能垒为16.66 kJ/mol的TS1 形成能量稳定的产物复合物CP1,CP1 进一步分解为产物CH2O、NO 和H2O。从CR1-TS1,C1-H3的键长伸长了0.009 nm,C1-H3 键断裂。OH 自由基的O 与cis-CH3ONO 的H 之间的距离由0.250 nm变为0.136 nm,最后在CP1 中O8-H3 键形成。在路径2 中,CR2 越过28.72 kJ/mol 的势垒高度形成过渡态TS2,cis-CH3ONO 中的C1-H4 键的键长伸长了0.012 nm。随着O8-H4 键的形成,cis-CH3ONO 的O6-N5 键断开,得到产物复合物CP2,形成的产物与路径1 相同。从能量来看,路径2 的反应势垒高于路径1,说明OH 自由基更容易与cis-CH3ONO甲基上对称面两侧的氢反应。
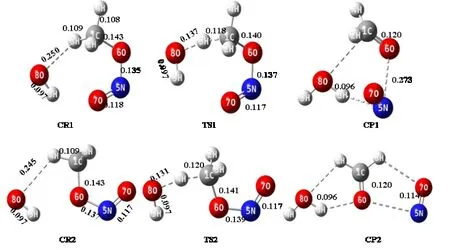
图2 cis-CH3ONO+OH 自由基发生氢提取反应的稳定构型Fig.2 Stable configuration of cis-CH3ONO+OH radical for hydrogen extraction reaction
2.1.2 OH 加成 如图3 和图4 所示,路径3和路径4 都是OH 自由基加成到cis-CH3ONO的N 原子上,形成相同的反应物复合物CR3(7.29 kJ/mol)。对于路径3,从CR3 到TS3 的反应势垒为31.48 kJ/mol。OH 自由基加成到cis-CH3ONO 后形成中间体CR4,O8-N5 已经成键。随着cis-CH3ONO 中N5-O6 单键的伸长,反应经过过渡态TS4 直接形成产物cis-HONO 和CH3O。TS4 相对于CR4 的势垒高度为28.42 kJ/mol,低于TS3 相对于CR3 的势垒高度(31.48 kJ/mol),所以路径3 中第一步是决速步。对于路径4,TS3a 相对于CR3 的势垒高度为30.85 kJ/mol,低于路径3 的第一步反应势垒。从TS3a 到CR4a,OH 自由基反向加成到cis-CH3ONO,O8-N5 键之间的距离逐渐缩短0.188~0.139 nm 并且成键。过渡态TS4a 相对于CR4a 的势垒高度为16.62 kJ/mol,O6-N5 键断裂得到产物trans-HONO 和CH3O。与路径3 一样,都是第一步为决速步,只是形成的产物不同。从反应势垒来看,路径4 的活化能低于路径3,说明OH 自由基加成到cis-CH3ONO 时更易生成 trans-HONO。

图3 cis-CH3ONO+OH 自由基发生OH 加成反应的稳定构型Fig.3 Stable configuration of cis-CH3ONO+OH radical with OH addition reaction obtained
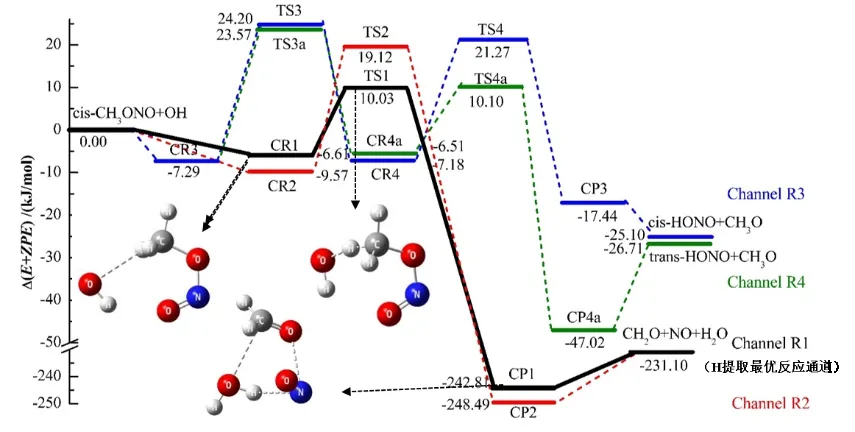
图4 cis-CH3ONO 与OH 自由基的反应势能面Fig.4 Potential energy surface of cis-CH3ONO reaction with OH·
从这四条反应路径的能量比较可以看出,产物的能量均低于反应物的能量,而且氢提取路径的反应势垒要低于OH自由基加成路径,说明在大气中cis-CH3ONO 与OH 自由基发生的是放热反应,反应机制主要为直接氢提取。
2.2 trans-CH3ONO 与OH 自由基的反应势能面
对于trans-CH3ONO+OH 自由基反应只存在两条反应路径,氢提取和OH自由基加成各一条,分别为路径5 和路径6。图5 显示了这两条反应路径中所有中间体优化后的分子构型及其结构参数,能量参数以及过渡态的虚频大小见表2,反应势垒图如图6 所示。
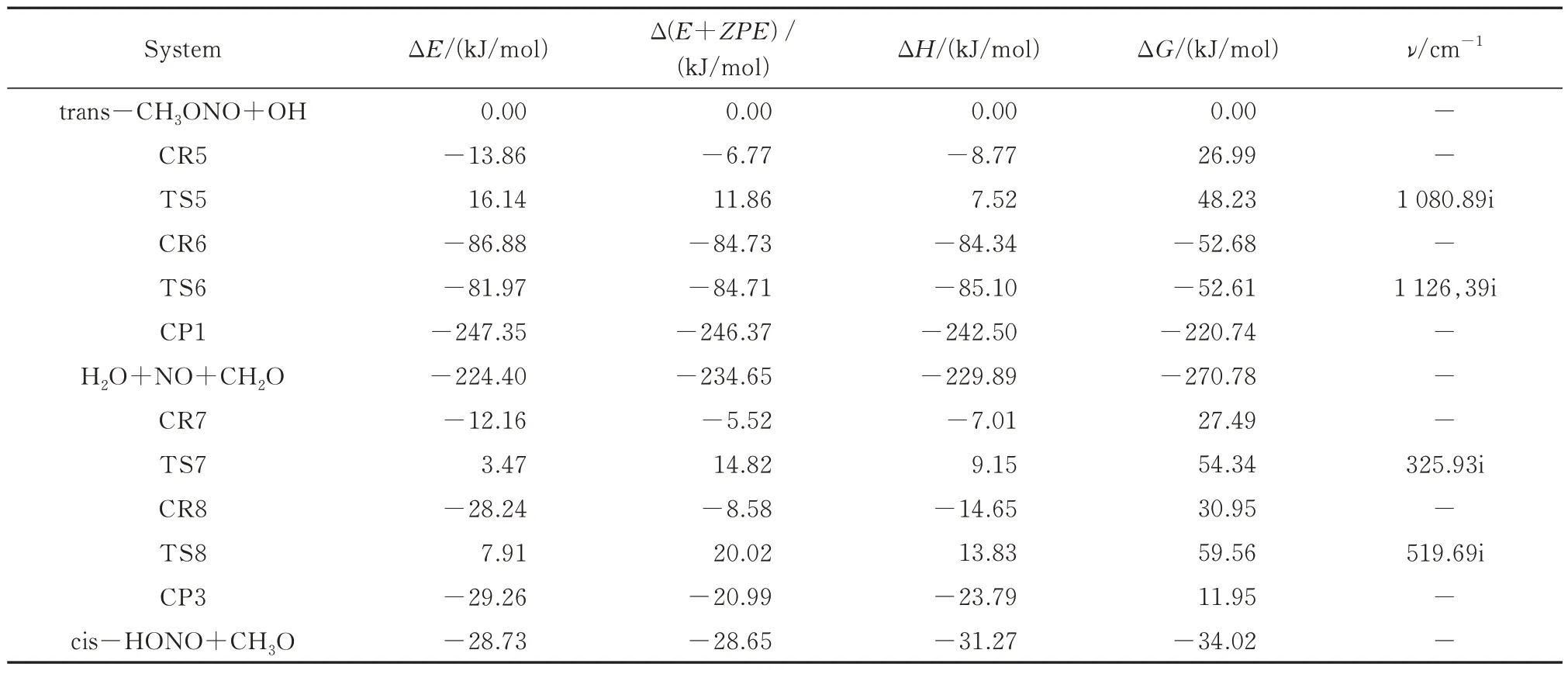
表2 trans-CH3ONO+OH 反应的能量参数及过渡态的虚频Tab.2 Energy parameters and virtual frequency of transition state of trans-CH3ONO+OH radical reaction
2.2.1 氢提取 如图5 和图6 所示,路径5 与路径1 相同都是OH 自由基中的O 直接提取CH3ONO 的H,但是路径5 需要经过两步反应才能形成产物复合物CP1。反应物复合物CR5 经过过渡态TS5 形成中间体CR6,TS5 相对于CR5 的能量为18.63 kJ/mol,比路径1 的反应势垒高0.087 kJ/mol。而CR6 的相对能量与TS6 相近,所以第二步反应可以实现无势垒转移,即O6-N5 键容易断裂形成NO 和CH2O。从能垒上来看,当CH3ONO 与OH 自由基发生H 提取反应时,cis-CH3ONO 比trans-CH3ONO 更容易发生。

图5 trans-CH3ONO+OH 自由基反应的稳定构型Fig.5 Stable configurations of trans-CH3ONO+OH radical reaction

图6 trans-CH3ONO 与OH 自由基的反应势能面Fig.6 Potential energy surface of trans-CH3ONO reaction with OH radical
2.2.2 OH 加成 trans-CH3ONO 与OH 自由基发生加成反应时,只存在一种加成方向。对于路径6,CR7 的相对能量为5.52 kJ/mol,比CR5 能量高。随着OH 自由基逐渐向trans-CH3ONO 靠近,CR7 经过能量为14.82 kJ/mol 的过渡态TS7 形成中间体CR8,此时OH 自由基已经加成到trans-CH3ONO 的N 原子上。trans-CH3ONO 中 的N5-O6 单键逐渐伸长,CR8 跨越28.59 kJ/mol 的反应势垒形成产物复合物CP3,进一步形成产物cis-HONO 和CH3O。在路径8 中,第二步的反应势垒高于第一步,因此第二步为决速步。与路径3 和4 的决速步对比,OH 自由基更易加成到trans-CH3ONO 上。对于trans-CH3ONO 与OH 自由基反应,同样是氢提取机制更容易发生,而且cis-CH3ONO 比trans-CH3ONO 更有利。
2.3 反应速率常数的计算
240~425 K 温度范围内CH3ONO+OH 自由基各反应路径的速率常数见表3,可以看出,反应速率常数与温度呈正相关,该结果与Nielsen 等[17]研究结果一致。路径1 的速率常数在298 K 下为1.07×10-14cm3·molecule-1· s-1,在cis-CH3ONO+OH反应四条路径中占主导地位。而trans-CH3ONO+OH自由基反应的主要路径为路径5,速率常数为1.89×10-15cm3· molecule-1· s-1,比cis-CH3ONO+OH自由基反应小一个数量级。因此在动力学上,同样cis-CH3ONO+OH 自由基反应比trans-CH3ONO+OH自由基反应更容易在大气中发生。在298K下,计算得到的CH3ONO+OH自由基的总速率常数为1.36×10-14cm3·molecule-1·s-1,比Nielsen[17]和Tuazon等[16]的结果小1个数量级。由于路径1的反应速率常数决定总反应速率常数的大小,因此为了确保结果的准确性,本文采用KiSThelp 程序重新计算了路径1 的反应速率常数,其结果为2.81×10-14cm3·molecule-1·s-1,与上述方法的结果近似。从目前的实验测定结果来看,Campbell[15]的实验结果与Nielsen[17]和Tuazon[16]之间的实验结果也相差10 倍,说明实验结果也不稳定。实验结果不一致可能与实验制取OH 自由基的来源有关,也可能与其测定反应速率的方法有关。本文的计算是根据第一性原理过渡态理论(TST)得到的反应机理,又根据过渡态理论及速率计算公式计算得到反应速率。理论计算不受OH 自由基来源、反应速率测定方法的影响,且用两种不同的速率计算方法得到了相近的计算结果,说明计算结果可靠,之所以出现理论与实验结果不一致,一方面理论计算有待加强,另一方面实验测定方法也有待改进,值得后续进一步验证。
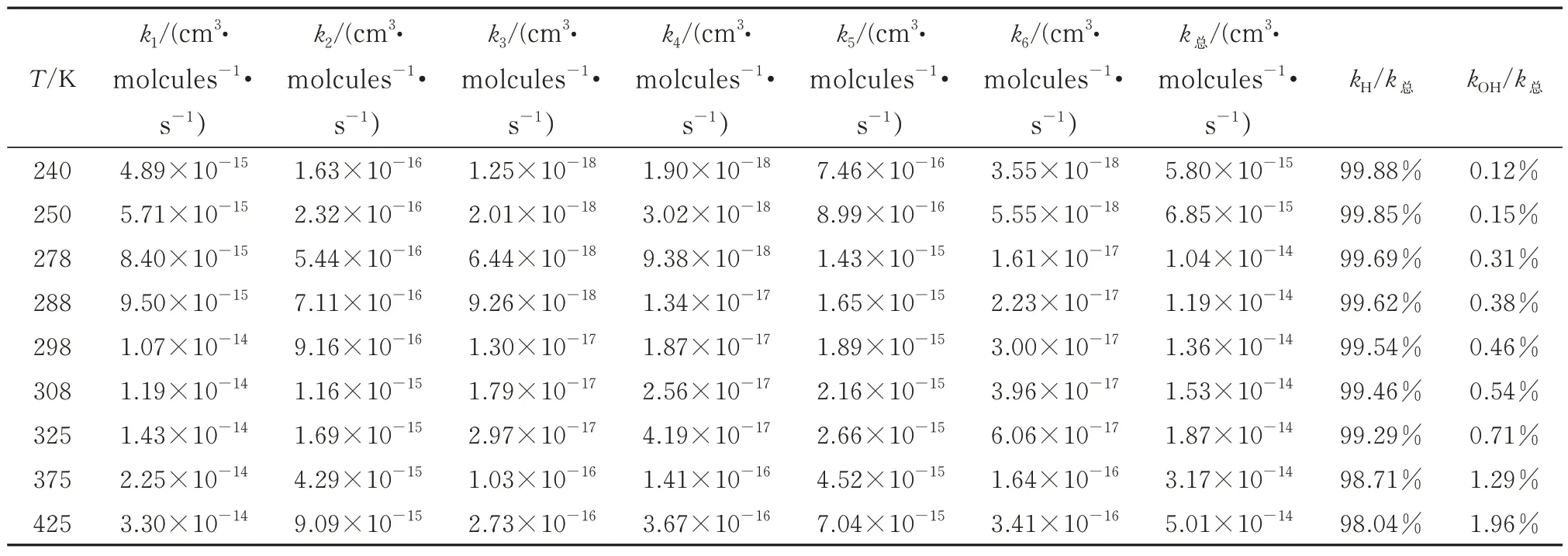
表3 240~425 K 温度范围内CH3ONO+OH 自由基反应速率常数Tab.3 Rate constants of CH3ONO+OH radical reaction at 240-425 K
氢提取路径和OH 加成路径反应速率随温度的变化如图7 所示。可以看到氢提取反应机制明显要优于OH 自由基加成反应机制。在240~425 K 温度范围内,二者反应速率常数相差2~3 个数量级。计算得到298 K 时H 提取路径和OH 自由基加成路径的反应速率常数比为kH/kOH=218.68,说明氢提取机制是CH3ONO 与OH 自由基反应的主要机制。从表3 中可以看到,随着温度的升高,kOH/k总的比值是增大的,即OH 自由基加成路径对总速率常数的贡献随温度增大而增大,但是与氢提取路径相比较,OH 自由基加成路径的贡献是可以忽略不计的。

图7 CH3ONO 与OH 自由基反速率常数与温度之间的关系Fig.7 Relationship between temperature and reverse rate constant of CH3ONO and OH radical
3 结论
CH3ONO 与OH 自由基反应共有六条反应路径,存在两种反应机制,即氢提取和OH 自由基加成,其中氢提取为主要反应机制。从反应势垒来看,cis-CH3ONO+OH 自由基反应比trans-CH3ONO+OH 自由基反应反应势垒低,更容易在大气中反应。通过对反应速率常数计算,发现cis-CH3ONO 最佳反应通道的速率常数比trans-CH3ONO 的大1 个数量级,H 提取路径的总反应速率常数要比OH 自由基加成大2~3 个数量级。尽管OH 自由基加成路径对总速率常数的贡献随温度增大而增大,但是与氢提取路径相比较,OH自由基加成路径的贡献是可以忽略不计的。
