基于催化的S-V反应含酰胺键聚合物的合成方法研究
2022-11-23刘伟,赵跃
刘 伟,赵 跃
(国网安徽省电力有限公司 电力科学研究院,安徽 合肥 230601)
“点击化学”在高分子科学和材料科学中发挥着越来越重要的作用。基于铜催化叠氮炔环加成反应(CuAAC)的点击化学概念由Sharpless等报道[1-3]。在2004年应用于合成聚合物化学后,应用CuAAC反应制备聚三唑端官能化聚合物和其他复杂大分子(后聚合)的研究也取得了成功[4-7]。受这一概念启发,其他点击反应,如硫醇烯反应、硫醇炔反应、Diels-Alder(D-A)反应和氨基炔反应已成功地引入聚合物化学中,用于制备含硫醚,分别是含乙烯基硫、含碳-碳或碳杂原子共轭和含烯胺的聚合物。然而,在某些情况下,过渡金属催化剂的使用、相对较高的反应温度或紫外光照射在一定程度上阻碍了它们的应用[8]。
含酰胺的化合物/聚合物在许多天然产物以及尼龙和芳酰胺等合成聚合物中非常普遍和重要[9]。酰胺形成的经典有机反应,包括胺与羧酸或活化衍生物的偶联反应,胺与羧酸的催化酰化,甲酰胺与胺的转酰胺,以及过渡金属(Ru、Au、Fe和Cu)催化醇或醛与胺的氧化偶联,均已得到很好的研究[10-12]。目前在高分子化学中只引入了几种传统的和新兴的酰胺键形成反应,但仍存在反应效率低、底物范围窄等问题。Vilarrasa等报道了催化Staudinger-Vilarrasa(S-V)反应,即在室温下使用2,2'-联吡啶二烯(PySeSePy)作为催化剂或活化剂,将羧酸与有机叠氮化合物直接酰胺化,并与上述传统的酰胺键形成反应进行了比较。催化S-V反应具有底物范围广、反应效率高、操作简便、反应条件温和、无金属等优点[13-15]。然而,尽管有机合成已经取得了很大进展,但这种有效的反应在聚合物化学中还没有实现。考虑到先前在聚合物科学中引入点击反应的成功,催化S-V反应与以往的点击化学反应(如CuAAC、巯基烯和D-A反应)具有相当的效率。
1 实验部分
1.1 材料
丙二酸、戊二酸、己二酸、壬二酸、癸二酸、新戊酸、琥珀酸、亚油酸、苯甲酸、对苯二甲酸:纯度均大于99.0%;2,2-双(4羧基苯基)六氟丙烷、硬脂酸、2-甲基己酸、1,3,5-苯三甲酸、1,12-二溴代癸烷、草酸、吡美利酸、1,18-十八烷二甲酸、聚乙二醇单甲醚、叠氮化钠(NaN3)、六甲基磷酸三胺(HMPA):纯度均大于98.0%;三甲基膦(Me3P,1.0 mmol/L甲苯溶液),茴香酸、胡椒酸、丙烯酸叔丁酯、2-溴吡啶、1,4-二(溴甲基)苯、丁二酸酐、1-甲基-2-吡咯烷酮(NMP)、溴化锂(LiBr):纯度均大于99.0%;苯乙烯(St):纯度大于99.5%;1-(4-甲氧基苯基)-环丙烷羧酸:纯度大于97.0%;4-二甲氨基吡啶(DMAP):纯度大于99.0%,用于真空显示;水合肼:纯度大于85.0%;盐酸体积分数为37%。
1.2 合成步骤
1.2.1二酸和二嗪催化S-V聚合的一般程序
将0.5 mmol/L二元酸、0.5 mmol/L二肼和0.2 mmol/L吡咯烷酮适量添加到50 mL反应管中,之后添加2.4 mL浓度为1.0 mol/L的Me3P甲苯溶液,温度为0 ℃,在手套箱中搅拌。将反应管加热到40 ℃气泡消失后24 h,加入3 mL水,搅拌30 min;然后加入适量水和甲醇,过滤、真空干燥过夜。粗产物经甲醇沉淀纯化。
1.2.2聚合物-N3端基与几种羧酸酰化反应的一般程序
在氮气气氛下,将浓度0.1 mmol/L的PS-N3适量、0.012 6 g浓度为0.04 mmol/L的PySeSePy和浓度为0.2 mmol/L的羧酸适量添加到配有磁力搅拌器的50 mL反应管中;然后将1.5 mL的Me3P溶液注入上述混合物中,温度为0 ℃;几分钟后,当不再观察到有氮气气泡时,将反应混合物在温度40 ℃条件下持续搅拌24 h。然后,在混合物中加入2~3 mL四氢呋喃,并将溶液沉淀到冷甲醇中。聚合物通过过滤获得,并在真空烘箱中于温度30 ℃条件下持续干燥24 h。PEG-N3端基的酰化反应过程与PS-N3类似,只是PEG-N3与羧酸反应,用冷无水乙醚作沉淀剂[16]。
1.2.3N3-聚合物-N3的端基与几种羧酸的酰化反应的一般程序
在氮气气氛下,将浓度为0.1 mmol/L的N3-PS-N3适量、0.025 1 g浓度为0.08 mmol/L的PySeSePy和浓度为0.4 mmol/L的羧酸适量加入装有磁力搅拌器的50 mL反应管中,之后将市售Me3P溶液(1.0 mmol/L甲苯溶液,3.0 mL,3.0 mmol/L)以0 ℃几分钟后,当不再观察到氮气气泡时,将反应混合物在温度40 ℃条件下持续搅拌24 h,然后在混合物中加入2~3 mL四氢呋喃,并将溶液沉淀到冷甲醇中。聚合物通过过滤获得,并在真空烘箱中于温度30 ℃条件下持续干燥24 h。
2 结果与讨论
2.1 S-V催化聚合聚酰胺的合成与表征
对于通过催化S-V反应进行的直接聚合,选择十四烷二酸和1,12-二氮杂十二烷作为模型单体来优化聚合条件。随着聚合时间的延长,聚合物的收率增加,24 h内达到94%。同时,所得聚酰胺的分子量达到约28 ku。假设酰胺键之间的氢键可能导致所得聚合物在甲苯中的溶解度相对较低,导致进一步的链生长停止[17]。此外,当PySeSePy的浓度从40 mmol/L提高到80 mmol/L时,所得聚合物的产率和分子量均降低,且多分散指数变宽。在此基础上,确定了最佳聚合条件:反应时间24 h,反应温度40 ℃,单体浓度为0.2 mol/L,Me3P浓度为1.0 mol/L。在此反应条件下,成功合成了一系列具有各种功能的聚酰胺。催化S-V聚酰胺化的合理机理,具体如图1所示。二嗪磷与Me3P反应生成Staudinger磷腈;同时,部分的二羧酸被PySeSePy激活生成二硒酯。然后,Staudinger磷腈与二硒醚反应生成中间产物N-PMe3SePy。中间体通过与剩余的二元酸反应迅速转化为聚酰胺和更多的二烯酸酯,这是整个聚合过程中的关键反应[18]。根据催化S-V聚酰胺化的可能机理,聚酰胺的端基可包括氨基或硒(NH2-PA-NH2,Py-Se-PA-Se-Py,NH2-Se-PA-Se-Py)。
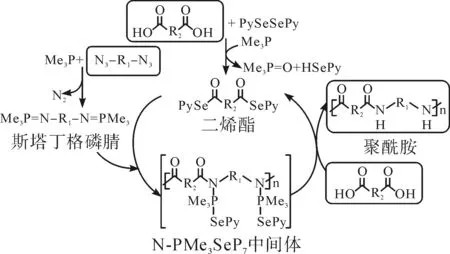
图1 催化S-V聚酰胺化的可能机理Fig.1 Possible mechanism for catalyzing the polyamideization of S-V
制备的聚合物的结构通过1H NMR、FT-IR、MALDI-TOF-MS光谱和SEC进行了表征,具体如图2、图3所示;所得聚合物PA1214用于结构表征。
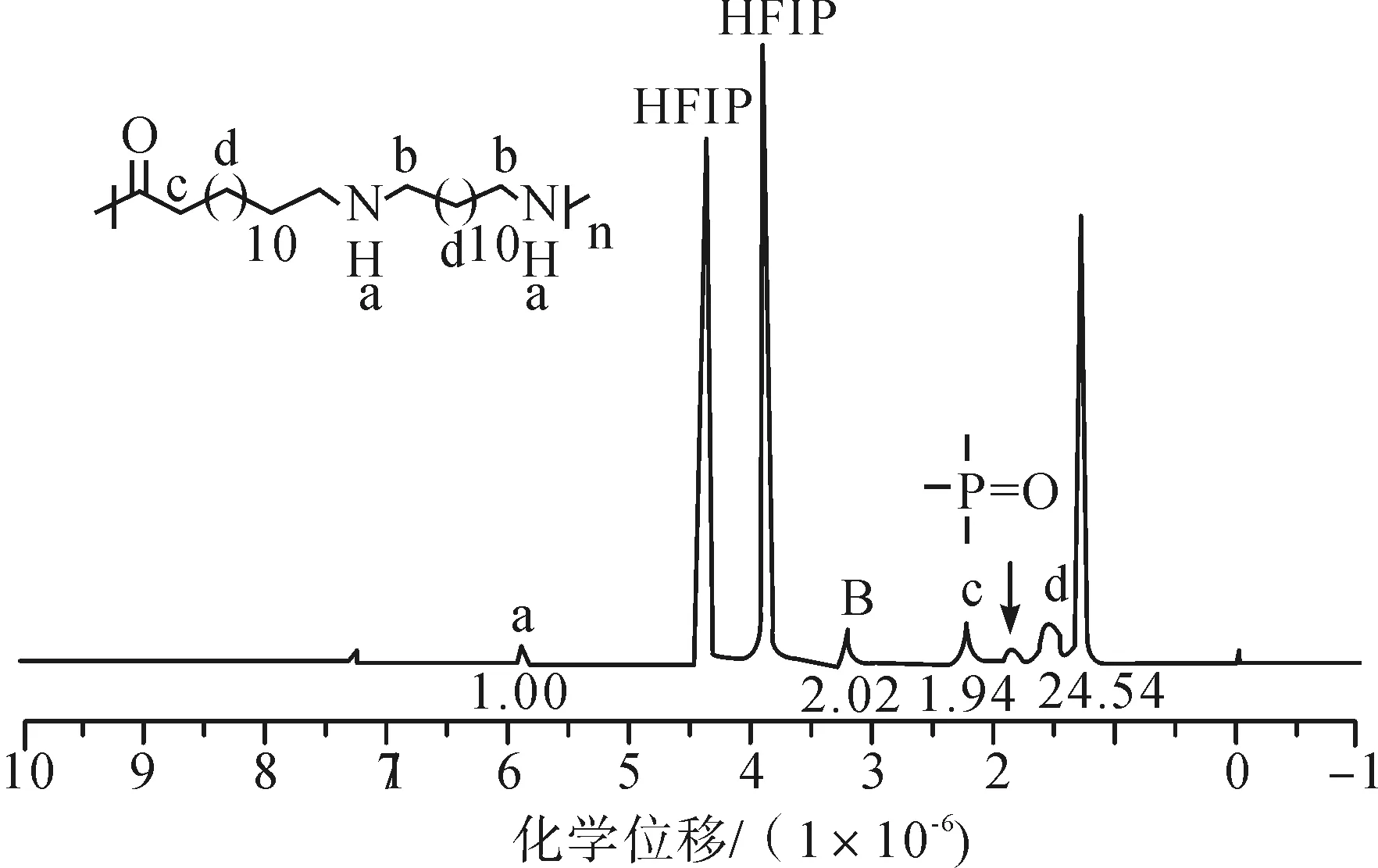
(a)

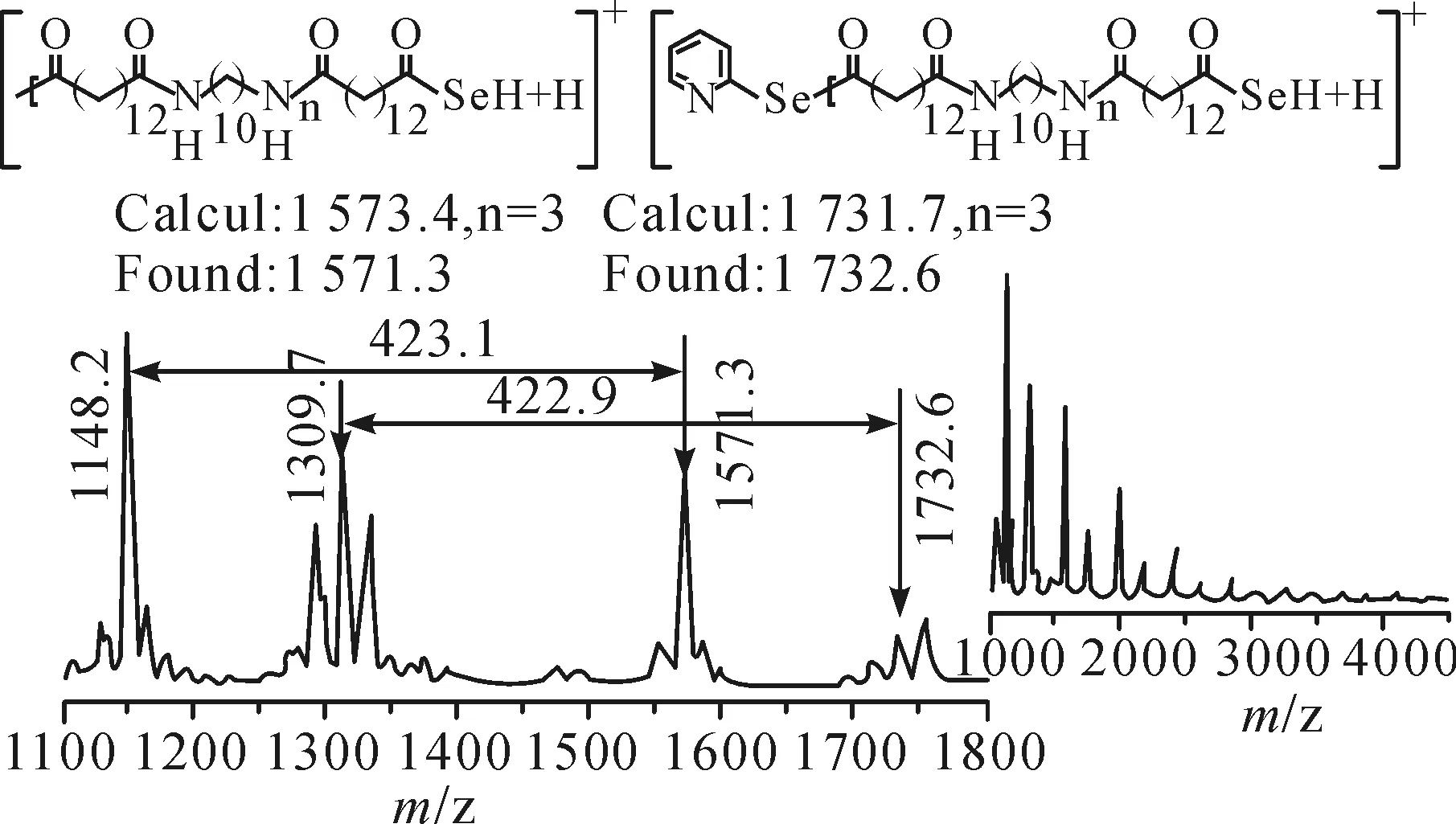
图3 PA1214的MALDI-TOF-MS光谱Fig.3 MALDI-TOF-MS spectrum of PA1214
由图3可知,当使用线性模式检测进行分析时,PA1214在MALDI-TOF-MS光谱中表现出2种不同的分布:一种是碎片离子[PA-SeH+H]+(实验质量=1 571.3 u,理论质量=1 573.4 u);另一种是碎片离子[PySe-PA-SeH+H]+(实验质量=1 732.6 u,理论质量=1 731.7 u)。这2个碎片离子是聚合物末端C—Se键断裂的结果,这些数据有力地支持了叠氮酸高效聚合的发生。
2.2 酰胺功能化(多)嵌段、星形和侧链聚合物的构建
聚酰胺和酰胺端官能化聚合物的成功制备启发我们构建更复杂的拓扑聚合物,如(多)嵌段、星形和侧链聚合物。以PEG-N3/PS-N3和二羧酸为起始组分,进行了叠氮链端基和酸基之间的S-V催化反应[19]。该高效偶联反应的效率通过1H NMR和SEC进行了表征,具体如图4所示。
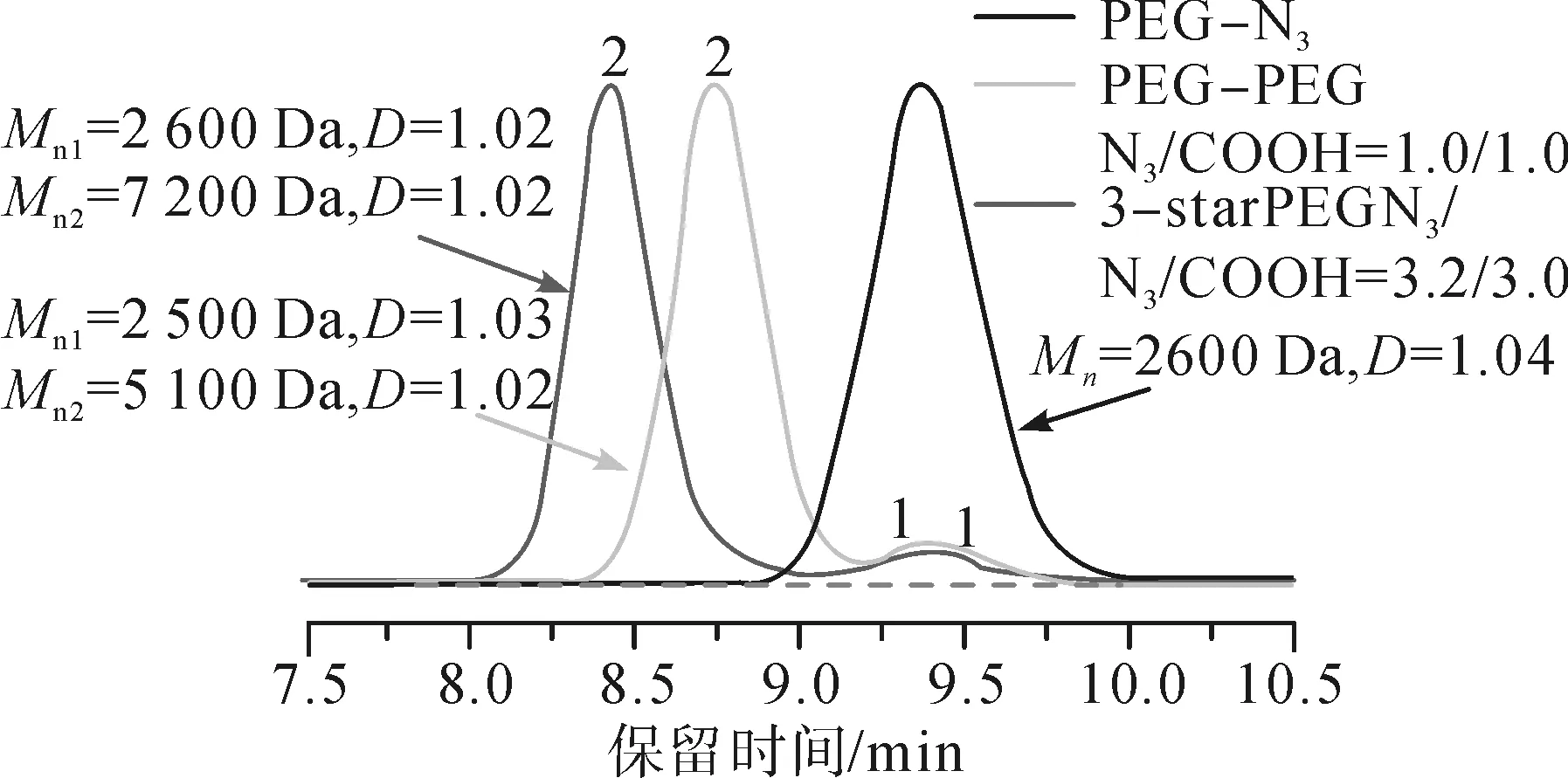
(a)
以PEG-N3与对苯二甲酸的模型反应为例讨论了偶联反应。预先制备的PEG-N3和与对苯二甲酸的偶联产物的1H NMR光谱分别如图5和图6所示。

图5 PEG-N3的1H NMR光谱(a) 制备的PEG-N3的1H NMR光谱(b)PEG-N3与胡椒酸S-V反应的酰胺官能化PEGCDCl3的CNMR谱Fig.5 1H NMR spectra of PEG-N3(a)1H NMR spectraof prepared PEG-N3 (b) CNMR spectra of amide functionalized PEG CDCl3 reacting PEG-N3 with piperic acid S-V
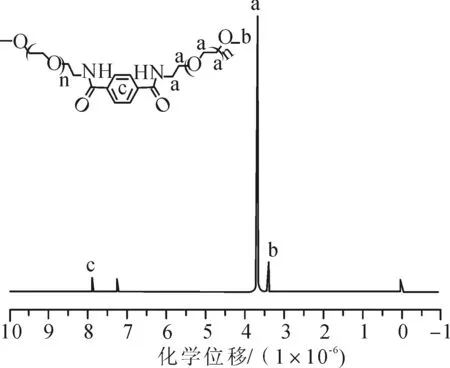
图6 PEG-N3与对苯二甲酸偶联反应产物的1H NMR谱图Fig.6 1H NMR spectra of the coupling reactionproducts of PEG-N3 and terephthalic acid
1H NMR谱中7.86×10-6~7.89×10-6处的新共振信号(见图5)归因于芳香环质子,表明芳香环的成功引入。偶联产物(PEG-PEG)的收率(质量分数)达(99±5)%的实验误差,这是由特征峰(图6中的芳香环质子c)和甲氧基质子(图6中的b)的相对峰面积估计的。此外,1H NMR估计的结果与使用高斯函数的偶联产物(PEG-PEG)SEC曲线的峰分裂结果(红色,见图4(a))很好地一致。SEC曲线(红色,图4(a))中偶联聚合物(PEG-PEG)的面积分数约为91.5%,其余的PEG链(8.5%的面积分数)被认为是在一个链端含有单酰胺基的PEG链或没有叠氮功能的PEG链的混合物。同时,观察到SEC轨迹明显向双分子量方向移动,这表明PS-PS的形成。这一结果证实了偶合反应对PS-N3也是高效的。
通过PEG-N3/PS-N3与偏苯三甲酸的高效偶联反应合成了3臂星形PEG和PS。预先制备的PEG-N3及其点击偶联产物的SEC曲线如图4(a)和图7所示。结果表明,当叠氮基与羧基的摩尔比为3.2∶3.0时,PEG-N3与偏苯三甲酸的高效偶联反应效率最高。在SEC曲线中,3臂星形PEG的面积分数约为94.9%(蓝色,图4(a)),略高于其他传统点击反应制备的面积分数。与初始PEG-N3样品具有相同保留时间的小峰可表示在链端含有单酰胺基的PEG链和不含叠氮官能团的PEG的混合物[20]。通过PEG-N3与偏苯三甲酸的S-V催化反应得到的三臂星形PEG的1H NMR光谱进一步证明了高效偶联反应的高效性。由图8可知,一个新的质子峰出现在8.48×10-6~8.50×10-6处。3臂星形PEG的百分比约为100%(带±5%的实验误差),这是由特征峰(图中的芳香环质子a)和甲氧基(图8中的c)的相对峰面积估计的。并且估计结果也与使用高斯函数的3臂星形聚合物的SEC曲线的峰分裂结果一致(蓝色,在图4(a)中),表明3臂星形聚合物以定量产率制备。
通过PEG-N3/PS-N3与偏苯三甲酸的高效偶联反应合成了3臂星形PEG和PS(方案S7b和S10)。预先制备的PEG-N3及其点击偶联产物的SEC曲线如图4a和图7所示。结果表明,当叠氮基与羧基的摩尔比为3.2∶3.0时,PEG-N3与偏苯三甲酸的高效偶联反应效率最高。在SEC曲线中,3臂星形PEG的面积分数约为94.9%(蓝色,图4a),略高于其他传统点击反应制备的面积分数。6c,25与初始PEG-N3样品具有相同保留时间的小峰可表示在链端含有单酰胺基的PEG链和不含叠氮官能团的PEG的混合物。通过PEG-N3与偏苯三甲酸的S-V催化反应得到的3臂星形PEG的1H NMR谱进一步证明了高效偶联反应的高效性。在图8中,一个新的质子峰出现在8.48×10-6~8.50×10-6(A)处。3臂星形PEG的百分比约为100%(带±5%的实验误差),这是由特征峰(图中的芳香环质子a)和甲氧基(图8中的c)的相对峰面积估计的。并且估计结果也与使用高斯函数的3臂星形聚合物的SEC曲线的峰分裂结果一致(蓝色,在图4a中),表明3臂星形聚合物以定量产率制备。
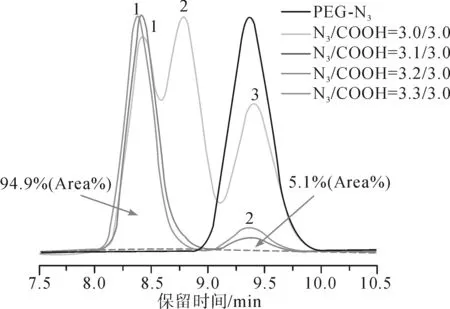
图7 PEG-N3及其与偏苯三甲酸偶联产物的SEC曲线Fig.7 SEC curve of prepared PEG-N3 and itscoupling product with trimellitic acid

图8 PEG-N3与偏苯三甲酸的偶联产物的1H NMR光谱Fig.8 1H NMR spectra of the coupling productof PEG-N3 and triphenyl trimeric acid
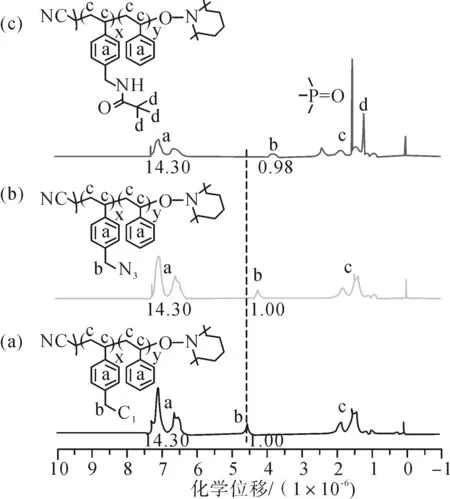
图9 侧链酰胺官能化PS(c)、预先制备的P(S-co-CMS-N3)(b)和P(S-co-CMS)(a)在25 ℃的1H NMR光谱CDCl3中的CFig.9 Side chain amide functionalized PS(c), pre-prepared P(S-co-CMS-N3)(b)and P(S-co-CMS)(a) in 1H NMR spectrum°CDCl3 at 25 ℃ c
由图9可知,1H NMR光谱清楚地显示了CMS亚甲基质子的共振峰从4.24×10-6移动到3.76×10-6(b,图9),证实了后聚合改性反应的完成。同时,1H NMR分析表明PS的侧链酰胺官能度含量高于98%。聚合改性后观察到的SEC痕迹明显向低分子量方向移动,这可能归因于聚合物链之间的氢键相互作用或聚合物链与SEC固定相之间的吸附相互作用。
3 结语
综上所述,首次成功地将催化S-V反应作为高分子科学中一种高效的化学策略。这种新的策略可以进一步有效地用于构建各种含酰胺的聚合物,包括通过直接点击聚合的聚酰胺、通过后聚合的酰胺官能化聚合物以及酰胺连接(多)嵌段共聚物和星形聚合物。最吸引人的一点,目前的策略可以很容易的在无金属的温和条件下操作,并且具有相对较高的效率,这可以与先前报道的那些突出的点击反应(CuAAC、巯基炔、巯基烯和D-a反应)相比较。这一策略为酰胺类聚合物的高效生产开辟了一条新的途径。值得注意的是,如果将来能够降低催化剂的负载量,这种化学作为点击策略的潜在用途是可行的。
