一例以皮肤色素沉着为唯一临床表现的X-连锁肾上腺脑白质营养不良的诊疗和基因检测分析
2022-11-21余佳瑜陈婷王智华郑涓曾天舒
余佳瑜,陈婷,王智华,郑涓,曾天舒
遗传资源
一例以皮肤色素沉着为唯一临床表现的X-连锁肾上腺脑白质营养不良的诊疗和基因检测分析
余佳瑜1,2,陈婷1,2,王智华1,2,郑涓1,2,曾天舒1,2
1. 华中科技大学同济医学院附属协和附属内分泌科,武汉 430022 2. 湖北省糖尿病与代谢疾病临床研究中心,武汉 430022
X-连锁肾上腺脑白质营养不良(X-linked adrenoleukodystrophy,X-ALD)是一种编码过氧化物酶体跨膜蛋白三磷酸腺苷结合盒亚家族D成员1(adenosine 5′-triphosphate binding cassette subfamily D member 1,)基因突变的遗传病,其具有多种临床表现,从首发症状到致死性炎性脱髓鞘的进展过程极其快速。因此,识别早期临床症状,早诊断、早治疗能有效阻止该疾病进展。本研究报道1例少见的以“皮肤色素沉着3年”为唯一临床表型的X-ALD患者的实验室检查及影像学特点,并采用高通量测序为患者及其父母进行基因测序。实验室检查结果提示患者肾上腺皮质功能减退,血清极长链脂肪酸浓度增高;肾上腺及脑MRI显示未见明显异常信号影。患者基因检测结果显示基因外显子1发生c.521A>C半合子突变,其母亲为同位点杂合突变,故诊断为“X-连锁肾上腺脑白质营养不良”。随访过程中,患者肾上腺皮质功能不全症状没有改善,颅脑MRI显示右侧放射冠、左侧顶叶脑白质见少许点状高flair信号影,提示可能出现脑损伤。仅有皮肤表现而无神经系统异常的X-ALD患者极易被忽视,早期诊断并给予早期干预是延缓该疾病进展的重要途径。因此,建议所有的男性儿童患者以皮肤色素沉着为唯一临床表现,继而诊断肾上腺皮质功能减退时,都应该进行基因检测,及早鉴别X-ALD。
X-连锁肾上腺脑白质营养不良;皮肤色素沉着;肾上腺皮质功能减退;基因
X-连锁肾上腺脑白质营养不良(X-linked adrenoleukodystrophy, X-ALD)是一种罕见的遗传性过氧化物酶体疾病,新生儿发病率约为1:14,700[1]。其主要病因是位于X染色体的ATP结合盒亚家族D成员1(ATP Binding Cassette subfamily D member 1,)基因突变,导致过氧化物酶体膜上的编码蛋白结构和功能障碍,超长链脂肪酸(very-long-chain fatty acid, VLCFA)跨膜转入过氧化物酶体降解失败,致使VLCFA异常沉积于血浆、神经系统和肾上腺等组织,从而出现中枢系统脱髓鞘和肾上腺功能不全为主的病理改变。因此,根据受累部位可将X-ALD分为4种类型:脑型(cerebral adrenoleukodystrophy, CALD)、肾上腺脊髓神经病型(adrenomyeloneuropathy, AMN)、单纯Addison型以及无症状型,单纯Addison型相对少见[2]。本病例以肾上腺皮质功能不全起病,以皮肤色素沉着为主要临床表现,通过总结该病例的特点,并对其后续治疗和病情进展进行随访分析,为诊疗临床表现不典型的X-ALD提供更多的临床证据。
1 对象与方法
1.1 研究对象与临床资料收集
患者,男,5岁6个月。因“皮肤变黑3年余”于2021年7月入住本院内分泌科,收集患者的病史、体格检查、实验室检查、影像学检查等临床资料。患者为独生子,父母均体健,无类似临床症状。由于亲属拒绝,无法获得其家系成员临床资料。对患者及其父母进行基因检测,向其说明基因测试的目的、程序、可能的结果、目前能够检测的范围,并保证保守秘密、尊重其隐私权。本研究获得华中科技大学同济医学院附属协和医院医学伦理委员会批准,患者监护人签署知情同意书。
1.2 外周血静脉采集与检测
采集患者外周静脉血用于多项临床生化指标检测。
促肾上腺皮质激素(ACTH)兴奋试验(3日静滴法):试验前检测基线8:00 am血皮质醇,24 h尿皮质醇。试验当天,排空膀胱后静脉持续8 h静滴ACTH 25IU,溶于5%葡萄糖500 mL中,静滴完成后抽取静脉血检测血皮质醇、次日8:00 am血皮质醇,并收集24 h尿测定尿皮质醇,连续3日。ACTH、皮质醇检测方法为罗氏电化学发光法。
VLCFA检测:采集患者外周静脉血后,经气相色谱质谱法测定血清极长链脂肪酸浓度。该检验由华中科技大学同济医学院附属同济医院遗传代谢性疾病诊断中心和广州金域医学检验中心完成。
1.3 全外显子组测序
采集患者及父母的肘前静脉血各2 mL,EDTA 抗凝以患者血液来源的基因组DNA为检测材料,首先将DNA打断并制备文库,然后通过Roche KAPA HyperExome芯片对目标基因外显子及邻近目标区的DNA进行捕获和富集,最后使用MGISEQ-2000测序平台进行变异检测。测序数据质控指标为:目标区域平均测序深度≥180×,其中目标区平均深度≥20×的位点所占比例>95%。测序片段通过BWA与UCSC hg19人类参考基因组进行比对,去除重复。使用GATK进行碱基质量值校正SNV、INDEL和基因型检测。使用ExomeDepth进行外显子水平的拷贝数变异检测。以上检测均由深圳华大基因检验实验室完成。
2 结果与分析
2.1 患者的临床表现
2.1.1 病史与体征
患者3年前无明显诱因出现口唇青紫、皮肤变黑,不伴发热,无呕吐,无呼吸困难,以“感冒”时青紫更为严重,运动时无明显加重,治疗未见好转;1年前皮肤变黑较前严重。半个月前本院儿科相关检查示:血随机皮质醇44.0 μg/L,24 h尿皮质醇3.6 μg/24 h(4.3~176.0 μg/24 h),24 h尿17-羟类固醇2.0 mg/24 h(2.0~10.0 mg/24 h),诊断为“肾上腺皮质功能不全”,为求进一步诊疗收治我科。患者足月顺产,体型健康,曾于11月龄有“抽搐”病史。查体:身高113 cm,体重19 kg,其身高体重位于同年龄、同种族和同性别儿童的中位数;心率90次/分,呼吸22次/分,血压98/69 mmHg;发育正常;全身皮肤色素沉着,以牙龈和唇尤为显著;神经系统查体示生理反射正常,腹壁反射存在,提睾反射存在,膝腱反射正常,巴宾斯基征阴性。
2.1.2 实验室检查结果
患者入院后实验室查体示:ACTH>2000.00 pg/mL (正常参考值:7~64 pg/mL)。ACTH兴奋试验结果如下:基线8:00 am血皮质醇40.0 μg/L(正常参考值:37.0~194.0 μg/L),24 h尿皮质醇3.6 μg/24 h(正常参考值:4.3~176.0 μg/24 h);第一日ACTH静滴后血皮质醇29 μg/L,次日8:00 am血皮质醇40 μg/L;24 h尿皮质醇3.0 μg/24 h(正常参考值:4.3~176 μg/24 h);第二日ACTH静滴后血皮质醇29 μg/L,次日8:00 am血皮质醇33 μg/L,24 h尿皮质醇2.9 μg/24 h;第三日ACTH静滴后血皮质醇33 μg/L,次日血皮质醇41 μg/L,24 h尿皮质醇2.2 μg/24 h。ACTH兴奋前后血、尿皮质醇均无明显上升,提示原发性肾上腺皮质功能减退。性激素、生长激素、胰岛素样生长因子、抗核抗体谱、降钙素原、C反应蛋白、免疫球蛋白、心肌损伤标志物、抗心肌抗体、凝血功能等均未见明显异常。
患者血浆VLCFA水平:C22:0 33.20 μmol/L(正常参考值:32.0±9.39 μmol/L),C24:0 54.83 μmol/L(正常参考值:27.5±9.17 μmol/L),C26:0 1.756 μmol/L(正常参考值:0.51±0.132 μmol/L),C26:0/C22:0 0.052(正常参考值:0.017±0.006),C24:0/C22:0 1.651(正常参考值:0.883±0.277)。C22:0正常,C24:0,C26:0,C26:0/C22:0,C24:0/C22:0均明显升高。
2.1.3 影像学检查结果
患者入院后脑平扫+增强核磁共振显示:脑实质未见明显异常信号影,脑室、脑沟裂未见明显增宽,中线结构居中,枕大池增宽,增强未见明显异常强化影。
肾上腺平扫+增强+三维显示双肾上腺形态大小及密度未见明显异常,增强未见明显异常强化肿块影。
2.2 基因测序结果分析
基因测序显示患者基因(NM_000033.3)外显子1发生c.521A>C半合子变异,导致第174位氨基酸由酪氨酸变异为丝氨酸,为错义突变。患者母亲一条性染色体上证明存在同位点的核苷酸杂合子变异,另一条性染色体为正常基因型。患者父亲未见上述变异(图1)。根据基因检测结果,患者更正诊断为“X-连锁肾上腺脑白质营养不良”。
2.3 治疗及随访
患者确诊“X-连锁肾上腺脑白质营养不良”后,予以醋酸泼尼松片1.25 mg/天对症治疗,以及洛伦佐油2~3 mL/kg/天治疗。
2.3.1 实验室检查结果
患者服用洛伦佐油治疗1个月后,随访复查血浆VLCFA水平:C22:0 42.4 nmol/mL(正常参考≤96.3 nmol/mL),C24:0 50.0 nmol/mL(≤91.4 nmol/mL),C26:0 1.93 nmol/mL(≤1.30 nmol/mL),C26:0/C22:0 0.046(≤0.023),C24:0/C22:0 1.18(≤1.39)。C22:0,C24:0,C24:0/C22:0正常,C26:0,C26:0/C22:0轻度升高。
患者确诊3个月后复查ACTH>2000 pg/mL,加大剂量为2.5 mg/天醋酸泼尼松片治疗;半年后复查ACTH仍大于2000 pg/mL,考虑到对患者身高的影响,改用氢化可的松每早10 mg下午5 mg,并嘱咐患者继续观察监测血ACTH水平。
2.3.2 影像学检查结果
患者确诊半年后,随访复查脑平扫+增强核磁共振,结果显示:双侧脑结构对称,灰白质对比正常,右侧放射冠、左侧顶叶脑白质见少许点状高flair信号影;各脑室、脑沟脑裂形态正常,中线结构居中,增强未见脑内明显异常强化影(图2)。
3 讨论
在X-ALD各分型中,儿童以炎性脑型最为常见,成人以AMN最为常见,单纯Addison型相对少见[3]。对单纯Addison患者随访观察后发现,几乎所有患者会随着疾病进一步发展成AMN甚至炎性脑脱髓鞘病[4]。一项前瞻性研究显示,约80%无症状患者有肾上腺功能不全相关生化指标的改变,其中70%的患者出现ACTH升高,这说明肾上腺功能不全的发生可能早于神经系统受累[5]。“单纯Addison”可能仅是患者出现神经系统症状的前期阶段[6]。因此,在X-ALD发病发展过程中,亚临床肾上腺功能不全和ACTH增高导致的皮肤色素沉着可能是唯一的早期临床表现。

图1 患者及其父母基因检测结果
患者(A)及其母亲(B)、父亲(C)基因测序序列图。箭头所示为突变位点。
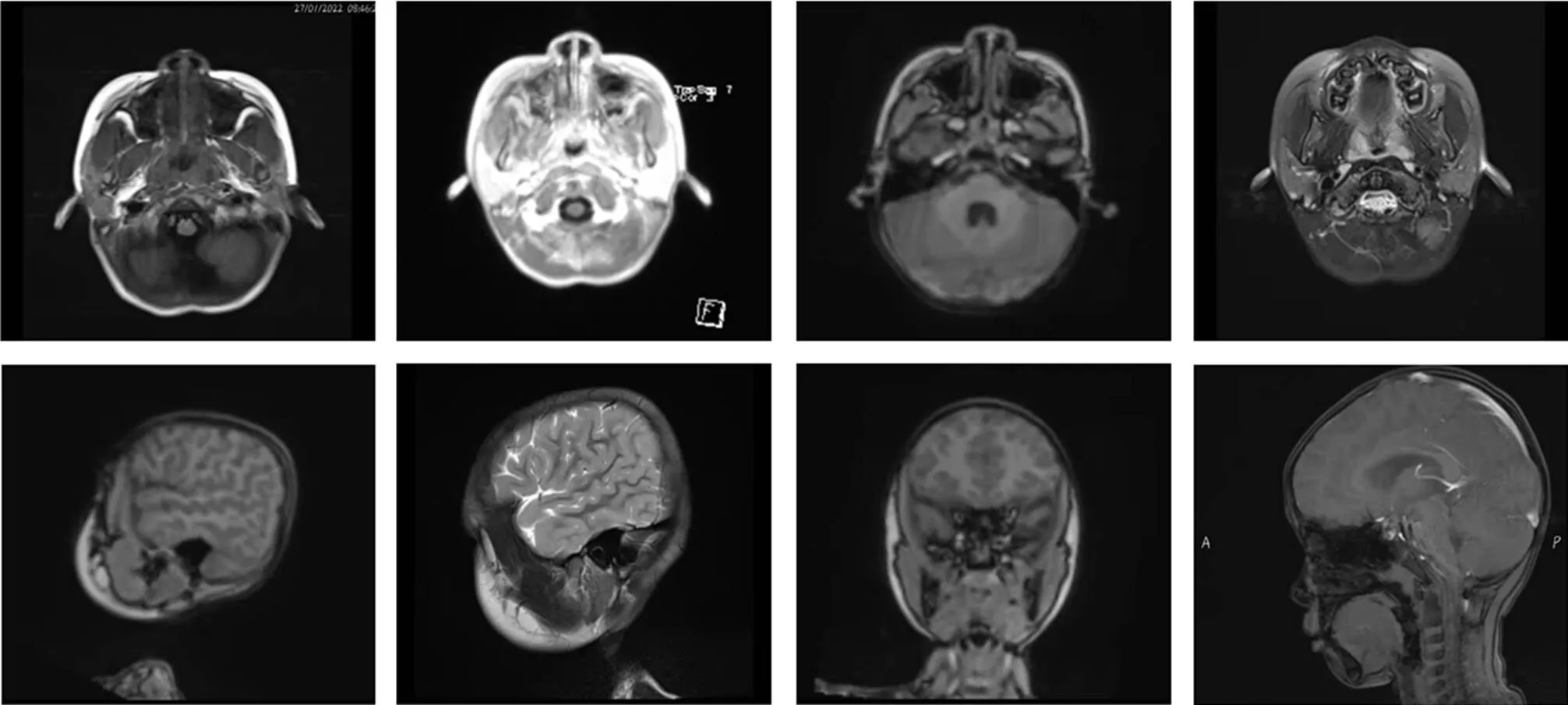
图2 患者脑MRI平扫+增强结果
右侧放射冠、左侧顶叶脑白质见少许点状高flair信号影。
本例患者以皮肤色素沉着为唯一临床表现,无乏力、恶心及呕吐等症状,排除自身免疫性肾上腺炎和感染性肾上腺炎等疾病,尽管其神经系统查体及脑MRI均正常,仍考虑遗传性疾病导致的肾上腺皮质功能减退,而基因检测也证实患者为X-连锁肾上腺脑白质营养不良。目前国内外报道的以皮肤色素沉着为唯一临床表现的病例较少[6],大多是色素沉着并发严重肾上腺皮质功能减退症状后才得以诊断和治疗[7],这是因为皮肤表现极其容易被忽视。然而,从单独的肾上腺功能不全进展到中枢系统脱髓鞘的过程是快速而致命的。因此,皮肤色素沉着作为可能的唯一首发症状,对有皮肤表现的患者进行肾上腺皮质功能检测及基因检测对X-ALD早期诊断和治疗至关重要。
目前基因已经发现3000余种基因突变,本例发现的变异位点521A>C共报道8例,其中7例为儿童脑型,1例为AMN。2例详细介绍病情过程,均以神经系统症状如行为异常或下肢痉挛起病[8,9],其中1例发作两次肾上腺危象但无皮素色素沉着[9]。本例患者病情与前述同变异位点病例的发病过程并不完全相似,其以肾上腺皮质功能不全起病,以皮肤色素沉着为唯一临床症状,并未发现神经系统表现。由于X-ALD病情存在不断进展的可能性,对该患者的疾病发展预测至关重要。然而目前缺乏基因突变型与临床表型相关的证据[10],所以相同突变位点的病情不能预测本例患者的疾病发展。因此,寻找X-ALD疾病进展的预测方法,并密切随访患者的病情状况,对早期防治有巨大意义。
VLCFA是X-ALD特异性诊断标志物,但其血浆浓度与疾病严重程度并不相关,无法预测疾病发展[11]。头颅核磁共振是目前最为主要的检测疾病严重程度与进展的方法,其能在神经系统症状发生前发现脑白质影像学改变,在疾病早期提示神经脱髓鞘和脑损伤[12]。因此,美国ALD无神经症状男童影像学检测指南与共识认为,对于X-ALD患者,尤其是男童,在进展成为炎性脑病之前,应进行连续脑MRI监测[13]。本例患者在确诊X-ALD后,立即进行脑MRI,显示无明显异常信号影;然而,在确诊后半年再次随诊复查脑MRI,提示右侧放射冠、左侧顶叶脑白质见少许点状高flair信号影。尽管患者目前无任何神经系统的症状,该结果仍然提示神经系统的受累,并且可能进一步发展成炎性脱髓鞘脑病。对于X-ALD患者,疾病的早期诊断和进展预测对其治疗至关重要,尤其是在未发展脑损伤的疾病早期阶段,对X-ALD患者进行神经功能量表和MRI严重程度评分以评估是否进行造血干细胞移植治疗,有望阻止炎性脑病的发生与进展[14]。
X-ALD目前没有很好的治疗手段,以改善症状为主[15]。对于肾上腺功能不全的X-ALD患者,美国ALD肾上腺检测指南建议ACTH大于300 pg/mL即需要糖皮质激素替代治疗,100~300 pg/mL的患者在兴奋试验后皮质醇仍小于18 μg/dL也考虑替代治疗[16]。有研究认为,考虑到对儿童身高的影响,应首选氢化可的松治疗,8~12 mg·m–2·d–1分3次口服;而达到终末身高后,可选用泼尼松或泼尼松龙1~2次/天,易于监测及简化用药[17]。为了防止肾上腺危象,应每3~6个月检测ACTH和皮质醇的变化趋势。此外,尽管盐皮质激素减少不常见,但也应该在诊断肾上腺功能不全后,每6个月监测血浆肾素活性和电解质等指标[16]。洛伦佐油的疗效仍有争议,本例患者在使用洛伦佐油后,尽管血浆VLCFA水平明显降低,然而对肾上腺皮质功能不全和神经系统损伤并无改善,甚至病情进行性加重。此前关于洛伦佐油的临床研究也发现,其既不能改善神经或内分泌症状,也不能阻止疾病进展[17],可能是因为洛伦佐油仅降低血浆中VLCFA水平,而不能大量进入大脑所致[18]。慢病毒造血干细胞移植的基因治疗在其2期临床试验中展现可靠的疗效[19],但是长期随访发现该疗法导致受试者骨髓异常增生综合征,所以其可行性仍需进一步商榷。造血干细胞移植是目前可能的唯一有效的治疗方式,能显著改善患儿的生存率[14]。然而,其局限于疾病早期未发生脑损伤阶段,且移植后远期疗效仍需进一步评估。因此,在无特效治疗方式的如今,X-ALD疾病的早诊断早发现尤为重要。
综上所述,本文报道了1例以皮肤色素沉着为唯一临床表现的X-ALD病例,对其诊治和随访过程进行详细介绍。38%的X-ALD患者肾上腺皮质功能不全是首发症状,且80%的患者终身肾上腺皮质受累。然而仅有皮肤表现,却无神经系统异常的X-ALD患者极易被忽视。早发现早诊断,尽早进行造血干细胞移植可能是目前延缓该疾病发展的唯一有效治疗手段。因此,所有的儿童男性患者以皮肤色素沉着为唯一临床表现,继而诊断肾上腺皮质功能减退,并排除免疫性和感染性肾上腺炎后,都应该进性基因检测,以鉴别X-ALD。
[1] Moser AB, Fatemi A. Newborn screening and emerging therapies for X-linked adrenoleukodystrophy., 2018, 75(10): 1175–1176.
[2] Kemp S, Huffnagel IC, Linthorst GE, Wanders RJ, Engelen M. Adrenoleukodystrophy-neuroendocrine pathogenesis and redefinition of natural history., 2016, 12(10): 606–615.
[3] Ashrafi MR, Amanat M, Garshasbi M, Kameli R, Nilipour Y, Heidari M, Rezaei Z, Tavasoli AR. An update on clinical, pathological, diagnostic, and therapeutic perspectives of childhood leukodystrophies., 2020, 20(1): 65–84.
[4] Engelen M, Kemp S, de Visser M, van Geel BM, Wanders RJA, Aubourg P, Poll-The BT. X-linked adrenoleukodystrophy (X-Ald): clinical presentation and guidelines for diagnosis, follow-up and management., 2012, 7: 51.
[5] Dubey P, Raymond GV, Moser AB, Kharkar S, Bezman L, Moser HW. Adrenal insufficiency in asymptomatic adrenoleukodystrophy patients identified by very long-chain fatty acid screening., 2005, 146(4): 528–532.
[6] Lee H, Ko JM, Lee SH. Generalized skin hyperpigmentationas the only manifestation of X-linked adrenoleucodystrophy., 2020, 182(1): 239–240.
[7] Nan M, Liu YF, Zhao X, Yuan N, Chai SB, Zhang XM. A pedigree of X-linked adrenoleukodystrophy with hypocorticism., 2021, 60(2): 152–155.
南敏, 刘玉芳, 赵心, 袁宁, 柴三葆, 张晓梅. 一个以肾上腺皮质功能减退起病的X连锁肾上腺脑白质营养不良家系报道. 中华内科杂志, 2021, 60(2): 152–155.
[8] Barceló A, Girós M, Sarde CO, Pintos G, Mandel JL, Pàmpols T, Estivill X.missense mutation Y174s in exon 1 of the adrenoleukodystrophy (Ald) gene., 1995, 95(2): 235–237.
[9] Horikawa Y, Enya M, Yoshikura N, Kitagawa J, Takashima S, Shimozawa N, Takeda J. A first case of adrenomyeloneuropathy with mutation Y174S of the adrenoleukodystrophy gene., 2017, 38(1): 13–18.
[10] Ping LL, Bao XH, Wang AH, Pan H, Wu Y, Xiong H, Jiang YW, Qin J, Wu XR. Clinical features and genotype-phenotype studies of 89 Chinese patients with X-linked adrenoleukodystrophy., 2007, 45(3): 203–207.
平莉莉, 包新华, 王爱花, 潘虹, 吴晔, 熊晖, 姜玉武, 秦炯, 吴希如. X连锁肾上腺脑白质营养不良89例临床特征及基因型/表型关系. 中华儿科杂志, 2007, 45(3): 203–207.
[11] Rattay TW, Rautenberg M, Söhn AS, Hengel H, Traschütz A, Röben B, Hayer SN, Schüle R, Wiethoff S, Zeltner L, Haack TB, Cegan A, Schöls L, Schleicher E, Peter A. Defining diagnostic cutoffs in neurological patients for serum very long chain fatty acids (Vlcfa) in genetically confirmed X-adrenoleukodystrophy., 2020, 10(1): 15093.
[12] Liberato AP, Mallack EJ, Aziz-Bose R, Hayden D, Lauer A, Caruso PA, Musolino PL, Eichler FS. MRI brain lesions in asymptomatic boys with X-linked adrenoleukodystrophy., 2019, 92(15): e1698–e1708.
[13] Mallack EJ, Turk BR, Yan H, Price C, Demetres M, Moser AB, Becker C, Hollandsworth K, Adang L, Vanderver A, Van Haren K, Ruzhnikov M, Kurtzberg J, Maegawa G, Orchard PJ, Lund TC, Raymond GV, Regelmann M, Orsini JJ, Seeger E, Kemp S, Eichler F, Fatemi A. Mri Surveillance of boys with X-linked adrenoleukodystrophy identified by newborn screening: meta-analysis and consensus guidelines., 2021, 44(3): 728–739.
[14] Mahmood A, Raymond GV, Dubey P, Peters C, Moser HW. Survival analysis of haematopoietic cell transplantation for childhood cerebral X-linked adrenoleukodystrophy: a comparison study., 2007, 6(8): 687–692.
[15] Yu JY, Chen T, Guo X, Zafar MI, Li HQ, Wang ZH, Zheng J. The role of oxidative stress and inflammation in X-link adrenoleukodystrophy., 2022, 9: 864358.
[16] Zhu J, Eichler F, Biffi A, Duncan CN, Williams DA, Majzoub JA. The changing face of adrenoleukodystrophy., 2020, 41(4): 577–593.
[17] Van Geel BM, Assies J, Haverkort EB, Koelman JH, Verbeeten B, Wanders RJ, Barth PG. Progression of abnormalities in adrenomyeloneuropathy and neurologically asymptomatic X-linked adrenoleukodystrophy despite treatment with "Lorenzo's Oil"., 1999, 67(3): 290–299.
[18] Rasmussen M, Moser AB, Borel J, Khangoora S, Moser HW. Brain, liver, and adipose tissue erucic and very long chain fatty acid levels in adrenoleukodystrophy patients treated with glyceryl trierucate and trioleate oils (Lorenzo's Oil)., 1994, 19(8): 1073–1082.
[19] Eichler F, Duncan C, Musolino PL, Orchard PJ, De Oliveira S, Thrasher AJ, Armant M, Dansereau C, Lund TC, Miller WP, Raymond GV, Sankar R, Shah AJ, Sevin C, Gaspar HB, Gissen P, Amartino H, Bratkovic D, Smith NJC, Paker AM, Shamir E, O'Meara T, Davidson D, Aubourg P, Williams DA. Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy., 2017, 377(17): 1630–1638.
Diagnosis, treatment and genetic analysis of a case of skin hyperpigmentation as the only manifestation with X-linked adrenoleukodystrophy
Jiayu Yu1,2, Ting Chen1,2, Zhihua Wang1,2, Juan Zheng1,2, Tianshu Zeng1,2
X-linked adrenoleukodystrophy (X-ALD) is an inherited disease caused by a mutation in the adenosine 5′-triphosphate binding cassette subfamily D member 1 () gene encoding a peroxisomal transmembrane protein, which has various clinical manifestations and a rapid progression from initial symptoms to fatal inflammatory demyelination. Therefore, identification of early clinical symptoms and further early diagnosis as well as treatment can effectively prevent disease development. In this study, we reported the laboratory and radiographic features in a rare case of X-ALD with 3-year skin hyperpigmentation as the only manifestation. And thegene was sequenced for the patient and his parents by a high-throughput sequencing method. The results of laboratory examination showed adrenocortical hypofunction and increased serum concentrations of very long-chain fatty acids. Brain MRI showed no obvious abnormal signal shadow. A hemizygous mutation of c.521A>C was detected in thegene of the patient, and his mother has the same site heterozygous mutation. Therefore, this patient was diagnosed as “X-linked adrenoleukodystrophy”.During the follow-up, adrenocortical hypothyroidism did not improve, and brain MRI showed few high-FLAIR signals in the white matter of the right radial corona and left parietal lobe, suggesting possible brain injury. X-ALD patients with only skin manifestations but no neurological abnormalities are easily neglected, but early diagnosis and early intervention are important ways to delay the progression of this disease.Therefore, genetic testing for early X-ALD is recommended in all male children patients with skin pigmentation as the sole clinical presentation and subsequent diagnosis of adrenal hypofunction.
X-linked adrenoleukodystrophy; skin hyperpigmentation; adrenocortical hypofunction;gene
2022-05-31;
2022-08-10;
2022-09-07
余佳瑜,在读硕士研究生,专业方向:内分泌与代谢。E-mail: yujiayu19970821@163.com
郑涓,博士,副教授,研究方向:内分泌与代谢。E-mail: zhengjuan25@163.com
曾天舒,博士,教授,研究方向:内分泌与代谢。E-mail: tszeng@126.com
10.16288/j.yczz.22-187
(责任编委: 周红文)
