2型糖尿病进程中胰岛β细胞功能变化的分子机制
2022-11-21吕承安王若然孟卓贤
吕承安,王若然,孟卓贤
综 述
2型糖尿病进程中胰岛β细胞功能变化的分子机制
吕承安1,2,王若然1,2,孟卓贤1,2
1. 浙江大学医学院病理学与病理生理学系,杭州 310058 2. 浙江大学医学院浙江省疾病蛋白质组学重点实验室,杭州 310058
近年来,2型糖尿病(type 2 diabetes,T2D)发病率迅速上升,已成为全球性的健康危机。最近的临床和基础研究表明,胰岛β细胞功能障碍是导致T2D及其相关并发症的重要原因。在2型糖尿病的自然病程中,胰岛β细胞经历从代偿到失代偿的动态变化;其中,代谢应激反应,如内质网应激(endoplasmic reticulum stress,ER stress)、氧化应激(oxidative stress)和炎症(inflammation)是β细胞功能变化的关键调控机制。本文总结了β细胞功能在2型糖尿病病程中动态变化的研究进展,以期深化对2型糖尿病分子机制的理解,为精准诊断和临床干预2型糖尿病提供参考。
胰岛β细胞;2型糖尿病;分子机制
2型糖尿病(type 2 diabetes,T2D)是当今世界上最严重的公共卫生问题之一,然而其具体发病机制仍不清楚,治疗效果远不理想[1]。T2D的主要特征是胰岛素分泌相对缺乏、外周组织胰岛素抵抗和随之而来的高血糖。长期高血糖将引起微血管和大血管并发症,导致心脏病、中风和糖尿病视网膜病变等严重疾病[2]。早期的研究曾认为外周组织胰岛素抵抗是T2D的主要病因;然而随着对胰岛生理功能和病理机制的深入了解,人们逐渐认识到胰岛β细胞(pancreatic β-cell)在T2D的发生发展过程中起到关键性作用[3]。全基因组关联研究(genome wide association study,GWAS)现已确定的T2D风险位点中,与β细胞功能受损相关的风险位点远多于与胰岛素抵抗相关的风险位点,进一步从遗传风险角度强调了胰岛β细胞在T2D中的核心作用[4]。
胰岛β细胞是人体血糖的主要调控者,通过对葡萄糖的精确感受分泌胰岛素,一方面促使外周组织摄取葡萄糖,另一方面抑制内源性葡萄糖生成,从而控制机体血糖稳态。在T2D的发病过程中,β细胞经历了从功能代偿(compensation)到失代偿(decompensation)的动态变化[5~7]。在糖尿病前期,虽然机体出现了轻度高血糖和外周组织胰岛素抵抗;但β细胞通过增加单个细胞胰岛素分泌量[7]和自我增殖等代偿反应克服这些障碍,维持了机体糖脂代谢稳态,延缓了糖尿病的发生[5,8]。然而,日积月累的代谢应激(metabolic stress),如内质网应激(endoplasmic reticulum stress,ER stress)、氧化应激(oxidative stress)和炎症(inflammation)会逐渐对β细胞造成损伤。随着这些因素的持续作用,β细胞代偿的能力逐渐减弱,最终发展为β细胞失代偿[7]。在失代偿阶段,β细胞分泌的胰岛素不足以满足身体的需求,机体糖脂代谢稳态被打破,糖尿病并发症逐一出现[5,8]。值得注意的是,除了代谢应激外,染色质重塑(chromatin remodeling)和组蛋白修饰(histone modification)也可能在胰岛β细胞功能从代偿到失代偿的转变中发挥关键作用[7];胰岛β细胞的去分化(dedifferentiation)和转分化(trans- differentiation)也在近年间被视为β细胞在应激条件下逃避细胞死亡命运的一种保护机制,与β细胞数量减少密切相关[9]。本文对胰岛β细胞动态变化机制的研究进展展开综述,讨论β细胞胰岛素分泌在生理状态下的调控,并进一步分别总结T2D前期促使β细胞代偿和晚期导致β细胞功能障碍和失代偿的分子机制。
1 β细胞胰岛素分泌的生理调节
β细胞正常合成和分泌胰岛素是机体调节血糖能力的重要保障。在正常情况下,胰岛素基因被转录,翻译为前胰岛素原(preproinsulin)并运输到内质网管腔;随后切除信号肽,剩余的肽链折叠并形成二硫键,形成胰岛素原(proinsulin);胰岛素原随后被转移到高尔基体,在高尔基体中被剪切为C肽(C-peptide)和胰岛素,然后被储存在分泌颗粒中,直到血糖的升高促使分泌颗粒与β细胞膜融合,胰岛素被释放进入血液循环[10]。
1.1 葡萄糖刺激胰岛素分泌通路
葡萄糖刺激胰岛素分泌(glucose-stimulated insulin secretion,GSIS)通路是发现最早且研究最透彻的胰岛素分泌通路。在GSIS通路中,血糖升高使β细胞膜葡萄糖转运体GLUT1(啮齿动物为GLUT2)将葡萄糖转运到胞内,随后葡萄糖激酶(glucose kinase, GK)将细胞内葡萄糖磷酸化为葡萄糖6-磷酸,这是β细胞中葡萄糖代谢的关键限速步骤。进一步在细胞质中进行糖酵解,产生丙酮酸(pyruvate)、还原性辅酶1(reduced form of nicotinamide-adenine dinucleotide,NADH)和少量腺苷三磷酸(adenosine triphosphate,ATP)。丙酮酸和NADH随后被运输到线粒体中,并通过三羧酸循环(tricarboxylic acid cycle)和电子传递链(electron transport chain,ETC),在此过程中产生大量的ATP;ATP转运复合物随后将这些ATP转运到细胞质中;细胞质中ATP/ADP比值的上升抑制了细胞膜上的ATP敏感钾离子(K+)的通道,并使细胞膜去极化;细胞膜去极化导致电压依赖性的钙离子(Ca2+)通道打开,胞外大量Ca2+流入;胞内Ca2+浓度的增加促进胰岛素的释放[10]。
由于被β细胞吸收的氨基酸和脂肪酸可以通过分解代谢形成三羧酸循环的中间产物,氨基酸和脂肪酸直接增强胰岛素分泌的作用可能可以部分归因于如上所述的钾通道-钙通道途径。例如,亮氨酸可以通过谷氨酸代谢途径产生α-酮戊二酸,然后进入三羧酸循环;同样,血液中的脂肪酸可以通过特殊的转运蛋白进入β细胞,被活化生成乙酰辅酶A(acetyl coenzyme A,acetyl-CoA),随后通过肉碱穿梭途径进入线粒体[11]。在线粒体基质中,乙酰辅酶A可被氧化,通过三羧酸循环和ETC产生ATP[11]。
1.2 胰岛素分泌的“放大通路”
除了经典的GSIS通路,氨基酸、脂肪酸以及胃肠道内分泌细胞分泌的肽类可以通过独立于GSIS通路的“放大通路”(amplifying pathways)来调控胰岛素的合成和分泌。虽然这些通路对β细胞胰岛素分泌功能有重要影响,但它们发现较晚,目前研究还不透彻。mTOR蛋白复合体1(mammalian target of rapamycin complex 1,mTORC1)对于β细胞感知氨基酸至关重要[12]。氨基酸可通过Rag和Rab1A蛋白激活β细胞的mTORC1,促进胰十二指肠同源框1(pancreatic and duodenal homeobox 1,PDX1)转录因子进入细胞核,提高胰岛素基因的表达水平[13]。G蛋白偶联受体40(G protein-coupled receptor 40,GPR40)是一种在β细胞表面高表达的脂肪酸受体;脂肪酸可结合并活化该受体,在胞内激活蛋白激酶C(protein kinase C,PKC),促使磷脂酰肌醇二磷酸(phosphatidylinositol diphosphate,PIP2)水解为三磷酸肌醇(triphosphoinositide,IP3)和二酰甘油(diacylglycerol,DAG);IP3结合并打开位于内质网膜上的IP3门控Ca2+通道,释放内质网中储存的Ca2+,增加胞浆Ca2+的浓度并促进胰岛素颗粒与细胞膜的融合。与此同时,DAG激活PKC和蛋白激酶D1 (protein kinase D1,PKD1),进而激发下游信号通路,导致胰岛素颗粒胞吐增强[14,15]。
抑胃肽(gastric inhibitory polypeptide,GIP)和胰高血糖素样肽1(glucagon-like peptide 1,GLP1)是两种重要的肠促胰激素,由肠道K细胞和L细胞分泌,对β细胞的正常分泌功能有重要作用。另外,生理条件下,胰岛中有少部分GLP1来源于胰岛α细胞[16]。GIP和GLP1与β细胞上的特定受体结合,介导腺苷酸环化酶(adenylate cyclase)将ATP转化为环磷酸腺苷(cyclic adenosine monophosphate,cAMP)。cAMP在胰岛β细胞中可以激活蛋白激酶A(protein kinase A,PKA)和Epac2。一方面,PKA抑制ATP敏感的钾通道,增加胰岛素分泌;另一方面,激活的Epac2诱导了内质网膜上的Ca2+通道RYR的开放,提升了胞内Ca2+的浓度[17]。也有研究表明,激活的Epac2可以与Rim2[18]、Piccolo和Rab3[19]等相互作用,诱导胰岛素的释放。
分泌到胞外的胰岛素可以通过自分泌的方式影响β细胞,对β细胞功能的正常发挥不可或缺[20]。胰岛素与β细胞膜上具有内在酪氨酸激酶特性的受体,如胰岛素受体(insulin receptor,IR)或胰岛素样生长因子-1受体(insulin-like growth factor 1 receptor,IGF-1R)结合,随后受体自磷酸化;激活的受体磷酸化胰岛素受体底物(insulin receptor substrate,IRS),IRS激活多个下游信号蛋白,包括Erk1/2、PI3K、Akt、mTORC1、p70s6K和PLC[20]。在生理条件下,胰岛素通过PI3K/p70s6K和CaMK等途径促进自身的转录[21]。此外,由于胰岛素可以通过PI3K-Akt通路激活mTORC1,mTORC1-PDX1介导的胰岛素表达也可能是胰岛素自分泌调节通路的另一个分支[20]。近期的研究进一步发现,胰岛素抑制受体Inceptor在胰岛素的自分泌调控中发挥着重要作用。β细胞表达的Inceptor可以促进网格蛋白介导的IR和IGF1R内吞,使得分泌出去的胰岛素不能激活自分泌调节通路;在小鼠中敲除Inceptor后,小鼠迅速出现了高胰岛素血症,并死于低血糖[22]。Inceptor的发现表明胰岛素自分泌反馈调节通路的负调控也具有重要的生理意义;适当抑制Inceptor,增强胰岛素自分泌调节可能是增强β细胞功能的新思路。
1.3 肠道菌群对β细胞胰岛素分泌的调控
作为人体内重要的微生态系统,肠道菌群对人体代谢、免疫、内分泌等具有深刻的影响,与T2D的关系十分密切[23,24]。近年来,多项研究证实了肠道菌群对胰岛β细胞功能的调控作用。肠道细菌受肠道溶菌酶作用产生的胞质肽聚糖可以进入血液循环,最终与β细胞膜上的受体Nod1结合,招募Rip2及Rab1a至β细胞内的分泌囊泡表面,促进胰岛素的成熟与转运;而在失去肠道菌群的无菌小鼠中,胰岛素的成熟和囊泡的正常转运过程受阻[25]。膳食纤维的摄入可以促进肠道内产短链脂肪酸(short-chain fatty acid,SCFA)细菌的繁盛;这些细菌产生的SCFA有助于促进β细胞分泌胰岛素[26,27]。一些研究把SCFA的这种生理效应归因于其对肠促胰激素GLP1的促进作用[26],也有一些研究认为某些SCFA是通过脑-肠轴刺激宿主的迷走神经,迷走神经促进β细胞分泌胰岛素[27]。此外,一种名为BefA的肠道细菌蛋白可以促进斑马鱼β细胞增殖[28]。这项研究深化了人们对肠-胰互作的认知,不过BefA在哺乳类和人类体内是否有相同的生理功能尚需进一步探究。
1.4 胰岛微环境对β细胞胰岛素分泌的调控
研究表明,在β细胞调控血糖的过程中,胰岛中的各种细胞,包括α细胞、δ细胞、免疫细胞以至胰岛神经细胞均发挥了作用。这样一个整合内分泌、免疫、神经调节因子的复杂系统被称作胰岛微环境[29]。胰岛内部的α细胞和δ细胞对β细胞具有重要的内分泌调节作用。α细胞产生的胰高血糖素(glucagon),δ细胞产生的生长抑素(somatostatin)等都可以通过旁分泌的形式作用于β细胞[30]。一方面,胰高血糖素可以促进β细胞分泌胰岛素;另一方面,胰高血糖素作用于δ细胞,刺激δ细胞产生生长抑素,对胰岛素的分泌起抑制作用;生长抑素反过来又抑制了胰高血糖素的分泌[30]。在这一经典模型之外,最近发现白细胞介素18(interleukin 18, IL18)也是β细胞功能的重要调控因子:IL18主要由α细胞分泌,可结合β细胞上的受体NCC,调控PDX1的表达,进而增强胰岛素合成与分泌[31]。免疫系统亦参与调节胰岛微环境。生理条件下,胰岛免疫细胞产生的炎症因子,如白细胞介素1β(interleukin 1β,IL1β)可以促进胰岛β细胞分泌胰岛素,参与维持餐后胰岛素分泌;IL1β信号缺失的小鼠出现了胰岛素分泌不足[32]。此外,神经对β胰岛也存在精细的调控。胰岛内部存在多种分泌不同神经递质的神经元[33],而它们分泌的神经递质对胰岛素分泌的影响不同:乙酰胆碱可以刺激胰岛素的分泌[34],而肾上腺素的生理效应则相反[35]。
2 T2D中胰岛β细胞的代偿机制
正常糖耐量个体中,胰岛素分泌量和胰岛素敏感性之间呈现出负相关:胰岛素敏感性低的个体需要更高的胰岛素分泌量才能实现正常的血糖控制。胰岛素分泌量的提升源自单个β细胞的胰岛素分泌水平提升和胰岛β细胞的数量提升[36]。β细胞增生的来源尚存在争议;许多研究表明胰岛α细胞[37]、δ细胞[38]以及胰腺导管细胞[39]均有转分化为β细胞的可能,也有研究表明生理和病理条件下新生的β细胞主要来源于已有β细胞的增殖[40]。经典理论认为代偿过程受β细胞对高糖的感知和响应调控;体内和体外实验均证明,在高糖刺激下,β细胞通过增大自身体积和增殖来增加胰岛素的合成和分泌量[41,42]。事实上,调控β细胞代偿的因素众多,这些因素的组合被统称为代谢应激[36]。β细胞代谢应激是由高血糖、高脂血症和高胰岛素血症介导的综合概念,由过多的能量摄入和外周组织胰岛素抵抗引起,主要包括内质网应激、氧化应激和炎症三大方面[43,44](图1)。β细胞代谢应激的来源十分广泛,涉及大脑、肝脏、肌肉、脂肪、肾脏和肠道等器官,也与胰岛的微环境有关[45]。
2.1 代偿期的内质网应激
胰岛素合成和分泌量的增加给β细胞带来了更重的蛋白质折叠任务,蛋白质折叠分子机器的超载最终导致未折叠及错误折叠蛋白质的大量积累[43],触发内质网应激。在高糖和外周胰岛素抵抗的情况下,高胰岛素需求刺激β细胞胰岛素的合成,并可能促进胰岛素的错误折叠[46]。高糖刺激显著增加了β细胞中内质网应激相关基因的表达,而过度刺激内质网应激通路可能是高糖对β细胞产生毒性的重要机制[47]。此外,内质网应激可能不仅由未折叠或错误折叠蛋白质积累引发:高脂则会刺激eIF-2磷酸化,诱导β细胞中ATF6、CHOP和BiP的表达,表明游离脂肪酸也参与了β细胞内质网应激[48,49]。有机制研究表明,饱和脂肪酸可能通过干扰和中断蛋白质从内质网到高尔基体的运输来触发内质网应激[50]。相反,高密度脂蛋白带来的脂质代谢改善恢复了内质网-高尔基转运稳态,缓解了β细胞内质网应激[51]。另外,棕榈酸诱导羧肽酶E(carboxypeptidase E,一种胰岛素原转化为成熟胰岛素所需的酶)的钙依赖性降解,从而导致胰岛素原的积累[52]。在过量的胰岛淀粉样多肽(islet amyloid polypeptide,IAPP)作用下,内质网应激反应也可以被激活[53,54]。
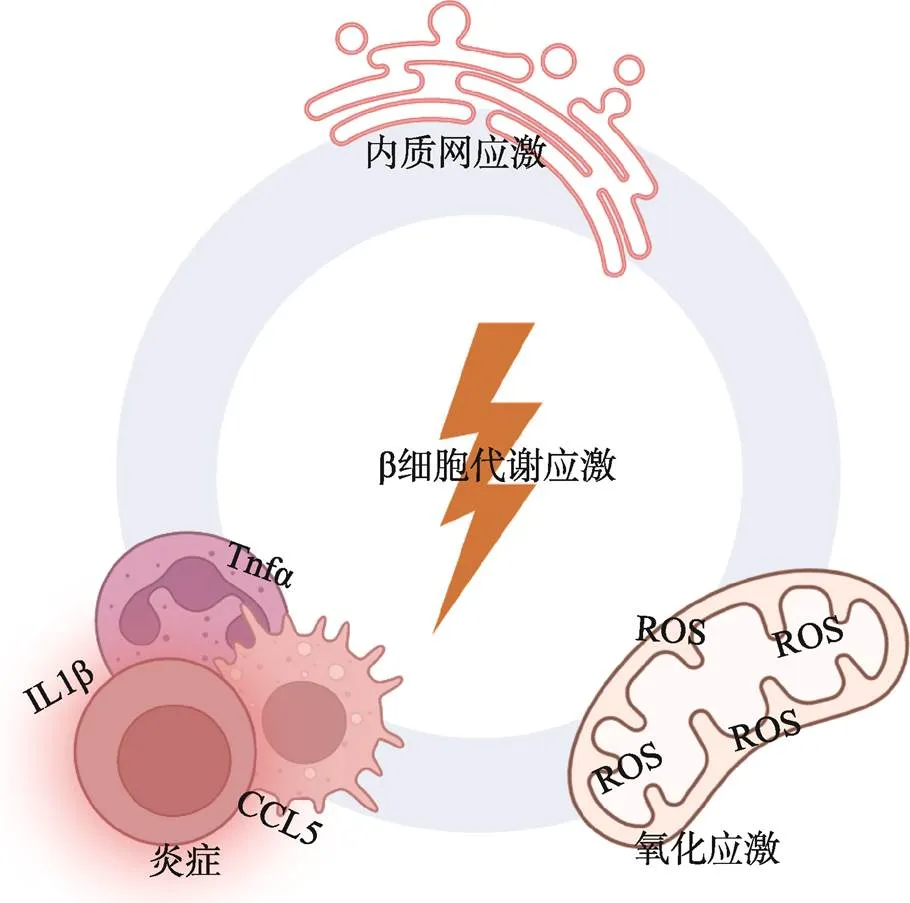
图1 胰岛β细胞代谢应激
三种主要的内质网信号分子,PERK、ATF6和IRE1α作为传感器触发细胞适应内质网应激的反应[55]。在β细胞代偿期内,内质网应激反应水平较低,持续时间较短,有助于维持蛋白质正常表达和折叠,包括抑制蛋白质翻译[56],诱导mRNA降解[57],激活自噬[58],促进内质网生成[59],上调伴侣蛋白表达水平[60]和降解错误折叠的蛋白[61]。总体来讲,代偿期的内质网应激反应从多方面保护β细胞内蛋白合成分子机器的正常运转,维持胰岛素的正常表达和分泌。代偿期的内质网应激对调控β细胞增殖也有重要意义。IRE1α可以通过调控转录因子XBP1的活性,促进细胞周期关键调控因子细胞周期素D1(cyclinD1)的转录表达;同时在内质网应激出现不久,IRE1α-XBP1信号通路即被激活,而IRE1α缺失,该条通路被抑制后,β细胞的代偿性增殖显著受损。这提示IRE1α-XBP1通路可能是代偿期β细胞增殖的重要调控因子[62]。也有研究证明ATF6亦有促进β细胞增殖的作用[63]。
2.2 代偿期的氧化应激
以活性氧(reactive oxygen species,ROS)水平升高为特征的氧化应激也是代偿期β细胞经历的一种代谢应激[60,64]。ROS是各种细胞生理活动的副产物,主要由线粒体有氧呼吸产生[65]。由于生理条件下β细胞的乳酸代谢水平很低[66],葡萄糖进入细胞时将被完全氧化;在高血糖条件下,如前所述,β细胞中糖氧化水平上升,同时ROS生成水平提高[64]。游离脂肪酸可能通过多种途径引起β细胞氧化应激。一方面,β-细胞中脂质氧化代谢的增加可能导致游离脂肪酸氧化不完全,导致ROS生成增加[64];另一方面,脂质介导的细胞氧化应激也可能与其抑制KIF12的作用有关[67]。KIF12是一种参与β细胞抗氧化活性的微管马达,可以作为转录因子Sp1的支架。KIF12-Sp1复合体通过提高Hsc70的表达,增强过氧化物酶体功能,从而缓解氧化应激[67]。同时,β细胞钙通道的持续激活和高胰岛素刺激都与其氧化应激水平有关[68,69]。
生理条件下,ROS作为信号分子,在信号转导中发挥着重要的作用[70]。在代偿期的氧化应激条件下,β细胞仍可以通过去除或中和过量的活性氧来自我保护。促进自噬,上调抗氧化酶和还原性抗氧化伴侣分子,如蛋白二硫异构酶(Protein disulfide isomerase,PDI),内质网氧化还原素1(endoplasmic reticulum oxidoreductins 1,ERO1)或谷胱甘肽(glutathione,GSH)等的表达[64,71]是β细胞对抗氧化应激的主要方法。在代偿阶段,β细胞内的抗氧化系统可以有效保护其免受ROS造成的种种损伤。Forkhead转录因子1(forkhead box O1,Foxo1)在β细胞强化抗氧化系统,应对氧化应激的过程中发挥了重要作用。在正常生理条件下,Foxo1可被Akt磷酸化,迁移到胞质并失活;而在氧化应激中,Foxo1被JNK等转录因子在另一位点磷酸化并入核,调控其下游基因表达,包括过氧化氢酶(catalase)和超氧化物歧化酶(superoxide dismutase,Sod2),发挥抗氧化作用[72]。β细胞也可能通过线粒体自噬以降解受损线粒体,从而维持线粒体稳态[73]。对健康小鼠的胰岛β细胞单细胞测序显示,ROS的产生可能可以通过下游的Srf、Jun、Fos等转录因子诱导β细胞增殖[74];提示ROS可能也在代偿期的β细胞增殖中发挥了重要作用。此外,也有研究认为ROS通过转录因子Nrf2参与β细胞代偿[75]。没有氧化应激时,Nrf2与抑制因子如Keap1结合,处于未激活状态,会被泛素化而降解;出现氧化应激时,Keap1被氧化使得对Nrf2的靶向结合能力下降,从而使Nrf2激活并转移至核内[75]。Nrf2一方面保护细胞免受ROS的损伤,抑制β细胞凋亡,另一方面促进β细胞增殖相关基因的表达[75]。
2.3 代偿期的胰岛炎症
糖尿病胰岛的炎症特征包括巨噬细胞浸润和炎症因子释放增加[76,77]。一方面,营养过剩刺激外周组织,如脂肪组织、肌肉和肝脏释放炎症因子进入血液循环,这些炎症因子随血流进入胰岛,导致胰岛炎症[78]。另一方面,营养过剩相关的高血糖和高血脂可以直接作用于胰岛,并触发炎症反应。相较于失代偿期,代偿期的胰岛炎症程度轻微。胰岛炎性细胞因子如IL1β、肿瘤坏死因子α(tumor necrosis factor α,TNFα)和干扰素γ(interferon γ,IFNγ)可以降低心肌肌浆网Ca2+-ATP酶(sarco endoplasmic reticulum Ca2+-ATPase,SERCA)的表达[79];由于SERCA的功能是将胞浆中的Ca2+转运到内质网中,这种调控可以在暂时将胞浆中的Ca2+维持在较高水平,从而促进胰岛素的分泌。此外,炎症因子还可通过调节Nrf2/NF-ΚB和SAPK/JNK通路诱导氧化应激[80],因此炎症和氧化应激的下游通路有可能存在交叉。在β细胞炎症和氧化应激通路的交互中,二价金属离子转运体1(divalent metal-ion transporter1,DMT1)可能发挥了重要作用[81]。
3 T2D中胰岛β细胞的失代偿机制
β细胞功能的代偿总是与不断增长的胰岛素需求相抗衡。后者的不断增长,连同T2D进程中其他因素,包括内质网应激、氧化应激和炎症水平的增加,使得β细胞的负担日益加重,最后导致代偿能力丧失,也即β细胞功能失代偿。可以说,β细胞的代偿和失代偿分别是由短期和长期的代谢应激的结果:短期的代谢应激会激发β细胞代偿反应;长期的代谢应激会对β细胞造成不可逆的损伤,包括凋亡和β细胞特性的丧失(去分化和转分化),是β细胞数量减少和功能损伤的直接原因(图2)。下面将对β细胞失代偿过程中三个方面的代谢应激和β细胞去分化、转分化的机制进行分别讨论。
3.1 失代偿期的内质网应激
在β细胞从代偿到失代偿的过渡过程中,胰岛素原的分泌显著增加,这表明β细胞中内质网或高尔基体中的胰岛素加工系统受损[60]。从已有的证据来看,β细胞从代偿到失代偿的转变可能对应于内质网应激反应从保护性到细胞毒性的转变。虽然代偿期的内质网应激具有一定的保护作用,失代偿期高水平、长期的内质网应激反应反而成为了β细胞凋亡的重要推手[55]。C/EBP同源蛋白(C/EBP homolog protein,CHOP)是PERK的下游因子,可能参与了内质网应激诱导的细胞凋亡。抑制CHOP基因可以缓解内质网应激[82],并减轻内质网应激后的β细胞凋亡;过表达CHOP则具有相反的作用[83]。此外,内质网应激可能通过干扰β细胞内质网膜上的钙离子通道,兰尼碱受体(ryanodine receptor,RyR)的功能,导致内质网钙离子渗漏[54];钙离子稳态的丧失导致胰岛素分泌功能受损,并进一步促进β细胞死亡。持久的内质网应激会减弱ATF6和IRE1α通路的活性,但PERK通路却不受影响[84],这意味着不同的应激反应通路在内质网应激压力下决定细胞命运方面发挥了不同的作用。结合上文对ATF6和IRE1α对β细胞的保护和促增殖效应的讨论[62,63],对这两条通路的抑制可能是失代偿期β细胞增殖功能减弱、凋亡增加的机制。
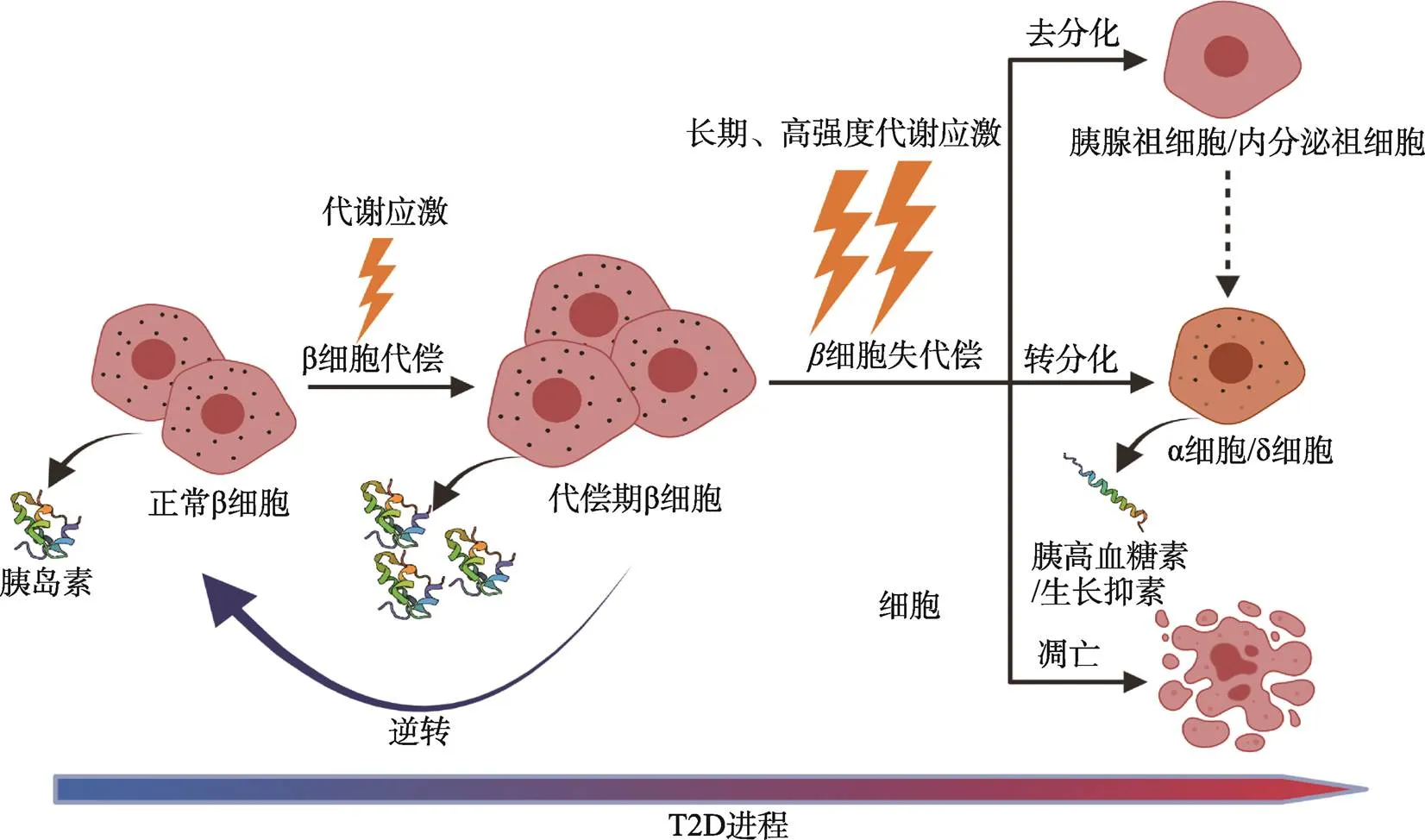
图2 T2D病程中胰岛β细胞从代偿到失代偿的功能变化
3.2 失代偿期的氧化应激
在病理条件下,过量的ROS会对细胞的核酸、蛋白质和脂质造成损伤,导致线粒体功能障碍,甚至诱导细胞死亡。正常的胰岛细胞会表达Sod2、catalase等过氧化物酶来清除过量的ROS,同时也会表达其他对氧化应激具有保护作用的因子,如Rheb1和Fam3a;然而在糖尿病个体的β细胞中,这些抗氧化酶和保护因子均出现了下调[85,86]。在失代偿期入核的Foxo1在长时间代谢应激下的耗竭可能参与助推β细胞抗氧化系统失效[72,87]。失代偿阶段,ROS过量,抗氧化系统受损,导致ROS破除β细胞的抗氧化防线,胞内的抗氧化剂耗竭[88]。此外,由于线粒体DNA(mitochondrial DNA,mtDNA)裸露在外,对ROS具有固有脆弱性,容易被氧化应激产生的ROS损伤。事实上,糖尿病患者常出现mtDNA损伤伴随线粒体功能障碍[89]。ROS还可能通过干扰线粒体形态损伤线粒体功能:体外研究表明,ROS抑制线粒体裂变的调节因子Fis1,并导致产生细长和巨大的线粒体[90]。联系Fis-1在β细胞功能中发挥的重要作用[91],提示ROS可能会通过抑制Fis-1导致异常的线粒体融合,进而损伤β细胞功能。综上,失代偿阶段发生的不可逆损伤,包括β细胞DNA损伤[92,93]和引发的细胞凋亡,可能是由ROS及其下游分子直接介导的。
3.3 失代偿期的炎症
生理情况下,炎症对维持β细胞功能发挥了重要作用;代偿期的炎症可以临时增加胰岛素分泌量;在失代偿期,过度的炎症反而促进β细胞功能损伤。虽然IL1β、TNFα和IFNγ等对SERCA的抑制作用可以短时间内增加细胞质钙的浓度,促进胰岛素分泌[79],长期暴露于这些炎症细胞因子之下会使得内质网钙离子储库耗竭,导致胰岛素分泌受损[79]。在这种情况下,Ca2+依赖的蛋白质加工和β细胞凋亡也会被促进[43]。胞外炎症因子的刺激之外,Eguchi等[94]的研究证明饱和游离脂肪酸可以通过TLR4/MyD88通路触发胰岛内的炎症过程,导致β细胞功能障碍,而抑制促炎激酶IKK可以部分抵消这种游离脂肪酸诱导的β细胞功能障碍[95]。炎症会激活氧化应激,而氧化应激则会刺激NF-κB通路,反过来加剧β细胞炎症[96]。2015年的另一项研究表明,炎症通过Nrf2/NF-κB和SAPK/JNK途径诱导ROS产生并促进β细胞死亡[80],表明促炎细胞因子引发的炎症反应与内质网应激和氧化应激密切相关。最新研究表明,炎症诱导β细胞死亡也需要胰岛免疫细胞的参与:炎症状态的胰岛中,胰岛内的巨噬细胞可与胰岛β细胞接触并诱导其增加中性粒细胞趋化因子Cxcl8a的表达;募集到的中性粒细胞攻击并杀死巨噬细胞接触过的β细胞[97]。
健康的肠道菌群可以维护β细胞的功能;反之,肠道菌群失调也是造成T2D中胰岛β细胞功能受损的因素[23]。目前的研究提示肠道菌群失调损伤胰岛β细胞功能的主要机制是加剧胰岛炎症。血液循环中对胰岛有促炎和损伤效果的细菌脂多糖(lipopolysaccharide,LPS)的增多与T2D患者出现的肠道菌群失调有关[98]。LPS会被免疫细胞识别,激活Toll样受体,通过NF-κB通路促进免疫细胞表达和分泌促炎性细胞因子[99]。另有研究表明,来源于肠道,含有细菌DNA的细胞外囊泡(microbial DNA- containing extracellular vesicles, mEV)在T2D患者的胰岛内富集;这些微囊泡以通过激活cGAS/STING导致炎症,进一步损伤β细胞功能[100]。mEV入侵胰岛可能与胰岛内Vsig4+巨噬细胞缺失有关[100];而mEV的产生与肠道菌群稳态之间的关系还不清楚,需要更深入的研究。
3.4 β细胞去分化与转分化
2012年,Talchai等[87]报道了T2D模型的小鼠胰岛中,高比例的β细胞分化程度出现了倒退,回到了前体细胞的状态,失去了胰岛素分泌的功能;这种丢失分化状态的独特生理学现象被命名为去分化。有研究显示T2D患者损失的β细胞大部分都是通过去分化途径,而不是通过凋亡途径损失的[101]。Foxo1对β细胞分化状态的维持具有重要的意义。在失代偿期,代偿期入核Foxo1的耗竭可能是β细胞去分化的重要推手:敲除Foxo1会导致只在前体β细胞中表达的基因,如Neurogenin3、Oct4、Nanog和L-Myc等在β细胞中表达,从而加速β细胞去分化[87]。
细胞谱系追踪实验表明,T2D患者的β细胞可能会转而分化为其他种类的细胞,如α细胞[102]和δ细胞[101];这种细胞命运的改变被命名为转分化[102]。发生这种转换的原因是在α细胞和δ细胞中特征性表达的基因在β细胞中被异常激活;如β细胞中Arx基因的激活会抑制β细胞特征性的Pax4基因,使其带有α细胞的特征[103,104]。目前尚不清楚β细胞转分化是一步到位抑或是需要经历去分化的中间阶段。虽然尚处在探索阶段,去分化和转分化现象的发现为T2D的干预和胰岛β功能的保护提供了新的思路。
4 结语与展望
β细胞在2型糖尿病中的动态变化在糖尿病的精准诊断和治疗中具有广阔的应用前景,已受到广泛的关注。但目前仍并没有技术能够准确地跟踪胰岛β细胞在人类糖尿病患者发病过程的纵向变化。这主要存在三方面的困境:第一是胰腺的解剖位置复杂,第二是具有内分泌功能的胰岛在胰腺中占比极低,第三是通过血液循环中胰岛素水平的变化来反映胰岛β细胞的功能,无法避免外周组织胰岛素敏感性对结果的干扰。基于啮齿动物模型,目前已经清楚β细胞在T2D中功能变化分为代偿和失代偿两个阶段(图2),但是尚没有找到这两个阶段之间的明确分界。鉴于有研究表明进入失代偿期前的β细胞仍有逆转的可能,探究这两个时期之间的界限,开发出相应的分子标志物,对糖尿病干预策略的制定和及时、精准的治疗可能大有裨益。
β细胞功能变化的调节可以在多个尺度上进行研究。T2D的进展会对多种器官组织产生复杂精细的影响,这些反过来又与β细胞发生深刻的相互作用,使β细胞不断调整自己的生理活动和分泌功能。与T2D进程中β细胞功能动态变化相关的有多种相互关联的途径,包括内质网应激、氧化应激、炎症及β细胞去分化、转分化。目前已有的药物大多将治疗策略定为增加β细胞的胰岛素分泌,长期用药势必使得β细胞超负荷工作,进一步加重其代谢应激,长远来看是一种竭泽而渔的做法;而对胰岛β细胞不同阶段的代谢特征和代谢应激通路的进一步探究将有助于新治疗靶点的发现和对应药物的开发,以β细胞代谢应激的逐渐缓解和功能的逐渐恢复为目的,为减轻T2D患者的胰岛β细胞功能损伤提供“治本”的方案。近期刊登于的研究靶向参与β细胞氧化应激的缺氧诱导因子-1α(hypoxia-inducible factor 1α,HIF1α),通过抑制HIF1α实现了β细胞代谢应激的缓解和功能的保护[105],是对这种新治疗思路的有效探索。近年来的新技术,诸如单细胞转录组学/表观基因组分析和基于CRISPR的基因组编辑等新技术可以在这方面为人们提供新的见解。鉴于单细胞转录组学/表观基因组技术目前只适用于新鲜组织,而大多数临床样本被固定包埋在石蜡中,因此开发固定后石蜡包埋或其他常见储存条件相兼容的新技术仍待解决[106]。
[1] Association AD. 2. Classification and diagnosis of diabetes: Standards of medical care in diabetes—2021. Diabetes Care, 2020, 44(Suppl 1): S15–S33.
[2] DeFronzo RA, Ferrannini E, Groop L, Henry RR, Herman WH, Holst JJ, Hu FB, Kahn CR, Raz I, Shulman GI, Simonson DC, Testa MA, Weiss R. Type 2 diabetes mellitus., 2015, 115019.
[3] Chiou J, Zeng C, Cheng Z, Han JY, Schlichting M, Miller M, Mendez R, Huang S, Wang JZ, Sui YH, Deogaygay A, Okino ML, Qiu YJ, Sun Y, Kudtarkar P, Fang RX, Preissl S, Sander M, Gorkin DU, Gaulton KJ. Single-cell chromatin accessibility identifies pancreatic islet cell type- and state-specific regulatory programs of diabetes risk., 2021, 53(4): 455–466.
[4] Krentz NAJ, Gloyn AL. Insights into pancreatic islet cell dysfunction from type 2 diabetes mellitus genetics., 2020, 16(4): 202–212.
[5] Hudish LI, Reusch JE, Sussel L. Beta cell dysfunction during progression of metabolic syndrome to type 2 diabetes., 2019, 129(10): 4001–4008.
[6] Hou J, Li Z, Zhong W, Hao Q, Lei L, Wang L, Zhao D, Xu P, Zhou Y, Wang Y, Xu T. Temporal transcriptomic and proteomic landscapes of deteriorating pancreatic islets in type 2 diabetic rats., 2017, 66(8): 2188–2200.
[7] Wang RR, Qiu XY, Pan R, Fu HX, Zhang ZY, Wang QT, Chen HD, Wu QQ, Pan XW, Zhou YP, Shan PF, Wang SS, Guo GJ, Zheng M, Zhu LY, Meng ZX. Dietary intervention preserves beta cell function in mice through ctcf- mediated transcriptional reprogramming., 2022, 219(7): e20211779.
[8] Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes., 2006, 116(7): 1802–1812.
[9] Zhang ZY, Gao Y, Meng ZX. Transcriptional control of pancreatic beta-cell identity and plasticity during the pathogenesis of type 2 diabetes., 2022, 49(4): 316–328.
[10] Fu Z, Gilbert ER, Liu DM. Regulation of insulin synthesis and secretion and pancreatic beta-cell dysfunction in diabetes., 2014, 9(1): 25–53
[11] Campbell JE, Newgard CB. Mechanisms controlling pancreatic islet cell function in insulin secretion., 2021, 22(2): 142–158.
[12] Jewell JL, Russell RC, Guan KL. Amino acid signalling upstream of mTOR., 2013, 14(3): 133–139.
[13] Zhang X, Wang XW, Yuan ZQ, Radford SJ, Liu C, Libutti SK, Zheng XFS. Amino acids-RAB1a-mTORC1 signaling controls whole-body glucose homeostasis., 2021, 34(11): 108830.
[14] Ferdaoussi M, Bergeron V, Zarrouki B, Kolic J, Cantley J, Fielitz J, Olson EN, Prentki M, Biden T, MacDonald PE, Poitout V. G protein-coupled receptor (GPR)40-dependent potentiation of insulin secretion in mouse islets is mediated by protein kinase D1., 2012, 55(10): 2682–2692.
[15] Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, Ogi K, Hosoya M, Tanaka Y, Uejima H, Tanaka H, Maruyama M, Satoh R, Okubo S, Kizawa H, Komatsu H, Matsumura F, Noguchi Y, Shinobara T, Hinuma S, Fujisawa Y, Fujino M. Free fatty acids regulate insulin secretion from pancreatic beta cells through gpr40., 2003, 422(6928): 173–176.
[16] Andersen A, Lund A, Knop FK, Vilsbøll T. Glucagon-like peptide 1 in health and disease., 2018, 14(7): 390–403.
[17] Holz GG. Epac: a new camp-binding protein in support of glucagon-like peptide-1 receptor-mediated signal transduction in the pancreatic beta-cell., 2004, 53(1): 5–13.
[18] Kashima Y, Miki T, Shibasaki T, Ozaki N, Miyazaki M, Yano H, Seino S. Critical role of camp-gefii—rim2 complex in incretin-potentiated insulin secretion., 2001, 276(49): 46046–46053.
[19] Ozaki N, Shibasaki T, Kashima Y, Miki T, Takahashi K, Ueno H, Sunaga Y, Yano H, Matsuura Y, Iwanaga T, Takai Y, Seino S. Camp-gefii is a direct target of camp in regulated exocytosis., 2000, 2(11): 805–811.
[20] Leibiger IB, Leibiger B, Berggren PO. Insulin signaling in the pancreatic beta-cell., 2008, 28: 233–251.
[21] Leibiger IB, Leibiger B, Moede T, Berggren PO. Exocytosis of insulin promotes insulin gene transcription via the insulin receptor/PI-3 kinase/p70 s6 kinase and cam kinase pathways., 1998, 1(6): 933–938.
[22] Ansarullah, Jain C, Far FF, Homberg S, Wißmiller K, von Hahn FG, Raducanu A, Schirge S, Sterr M, Bilekova S, Siehler J, Wiener J, Oppenländer L, Morshedi A, Bastidas-Ponce A, Collden G, Irmler M, Beckers J, Feuchtinger A, Grzybek M, Ahlbrecht C, Feederle R, Plettenburg O, Müller TD, Meier M, Tschöp MH, Coskun Ü, Lickert H. Inceptor counteracts insulin signalling in β-cells to control glycaemia., 2021, 590(7845): 326–331.
[23] Qin JJ, Li YR, Cai ZM, Li SH, Zhu JF, Zhang F, Liang SS, Zhang WW, Guan YL, Shen DQ, Peng YQ, Zhang DY, Jie ZY, Wu WX, Qin YW, Xue WB, Li JH, Han LC, Lu DH, Wu PX, Dai YL, Sun XJ, Li ZS, Tang AF, Zhong SL, Li XP, Chen WN, Xu R, Wang MB, Feng Q, Gong MH, Yu J, Zhang YY, Zhang M, Hansen T, Sanchez G, Raes J, Falony G, Okuda S, Almeida M, LeChatelier E, Renault P, Pons N, Batto JM, Zhang ZX, Chen H, Yang RF, Zheng WM, Li SG, Yang HM, Wang J, Ehrlich SD, Nielsen R, Pedersen O, Kristiansen K, Wang J. A metagenome-wide association study of gut microbiota in type 2 diabetes., 2012, 490(7418): 55–60.
[24] Tremaroli V, Bäckhed F. Functional interactions between the gut microbiota and host metabolism., 2012, 489(7415): 242–249.
[25] Zhang Q, Pan Y, Zeng BH, Zheng XJ, Wang HF, Shen XY, Li H, Jiang Q, Zhao JX, Meng ZX, Li PP, Chen ZJ, Wei H, Liu ZH. Intestinal lysozyme liberates Nod1 ligands from microbes to direct insulin trafficking in pancreatic beta cells., 2019, 29(7): 516–532.
[26] Zhao LP, Zhang F, Ding XY, Wu GJ, Lam YY, Wang XJ, Fu HQ, Xue XH, Lu CH, Ma JL, Yu LH, Xu CM, Ren ZY, Xu Y, Xu SM, Shen HL, Zhu XL, Shi Y, Shen QY, Dong WP, Liu R, Ling YX, Zeng Y, Wang XP, Zhang QP, Wang J, Wang LH, Wu YQ, Zeng BH, Wei H, Zhang MH, Peng YD, Zhang CH. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes., 2018, 359(6380): 1151–1156.
[27] Perry RJ, Peng L, Barry NA, Cline GW, Zhang DY, Cardone RL, Petersen KF, Kibbey RG, Goodman AL, Shulman GI. Acetate mediates a microbiome-brain-β-cell axis to promote metabolic syndrome., 2016, 534(7606): 213–217.
[28] Hill JH, Franzosa EA, Huttenhower C, Guillemin K. A conserved bacterial protein induces pancreatic beta cell expansion during zebrafish development., 2016, 5: e20145.
[29] Almaça J, Caicedo A, Landsman L. Beta cell dysfunction in diabetes: The islet microenvironment as an unusual suspect., 2020, 63(10): 2076–2085.
[30] Huising MO. Paracrine regulation of insulin secretion., 2020, 63(10): 2057–2063.
[31] Zhang X, Luo SY, Wang MJ, Huang Q, Fang WQ, Li J, Liu TX, Zhang YY, Deng ZY, Liu CL, Guan SL, Ayala JE, Flavell RA, Kulkarni RN, Libby P, Guo JL, Liu ZS, Shi GP. IL18 signaling causes islet β cell development and insulin secretion via different receptors on acinar and β cells., 2022, 57(12): 1496–1511.e1496.
[32] Dror E, Dalmas E, Meier DT, Wueest S, Thévenet J, Thienel C, Timper K, Nordmann TM, Traub S, Schulze F, Item F, Vallois D, Pattou F, Kerr-Conte J, Lavallard V, Berney T, Thorens B, Konrad D, Böni-Schnetzler M, Donath MY. Postprandial macrophage-derived IL-1β stimulates insulin, and both synergistically promote glucose disposal and inflammation., 2017, 18(3): 283–292.
[33] Thorens B. Neural regulation of pancreatic islet cell mass and function., 2014, 16(Suppl 1): 87–95.
[34] Gilon P, Henquin JC. Mechanisms and physiological significance of the cholinergic control of pancreatic beta-cell function., 2001, 22(5): 565–604.
[35] Rosengren AH, Jokubka R, Tojjar D, Granhall C, Hansson O, Li DQ, Nagaraj V, Reinbothe TM, Tuncel J, Eliasson L, Groop L, Rorsman P, Salehi A, Lyssenko V, Luthman H, Renström E. Overexpression of alpha2A- adrenergic receptors contributes to type 2 diabetes., 2010, 327(5962): 217–220.
[36] Lv CA, Sun YC, Zhang ZY, Aboelela Z, Qiu XY, Meng ZX. β-cell dynamics in type 2 diabetes and in dietary and exercise interventions., 2022, doi: 10.1093/jmcb/mjac046.
[37] Thorel F, Népote V, Avril I, Kohno K, Desgraz R, Chera S, Herrera PL. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss., 2010, 464(7292): 1149–1154.
[38] Chera S, Baronnier D, Ghila L, Cigliola V, Jensen JN, Gu G, Furuyama K, Thorel F, Gribble FM, Reimann F, Herrera PL. Diabetes recovery by age-dependent conversion of pancreatic δ-cells into insulin producers., 2014, 514(7523): 503–507.
[39] Gribben C, Lambert C, Messal HA, Hubber EL, Rackham C, Evans I, Heimberg H, Jones P, Sancho R, Behrens A. Ductal Ngn3-expressing progenitors contribute to adult β cell neogenesis in the pancreas., 2021, 28(11): 2000–2008.e2004.
[40] Zhao H, Huang XZ, Liu ZX, Pu WJ, Lv Z, He LJ, Li Y, Zhou Q, Lui KO, Zhou B. Pre-existing beta cells but not progenitors contribute to new beta cells in the adult pancreas., 2021, 3(3): 352–365.
[41] Zhang HJ, Zhang J, Pope CF, Crawford LA, Vasavada RC, Jagasia SM, Gannon M. Gestational diabetes mellitus resulting from impaired beta-cell compensation in the absence of FoxM1, a novel downstream effector of placental lactogen., 2010, 59(1): 143–152.
[42] Cerf ME. High fat programming of beta cell compensation, exhaustion, death and dysfunction., 2015, 16(2): 71–78.
[43] Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus., 2008, 29(1): 42–61.
[44] Nordmann TM, Dror E, Schulze F, Traub S, Berishvili E, Barbieux C, Böni-Schnetzler M, Donath MY. The role of inflammation in β-cell dedifferentiation., 2017, 7(1): 6285.
[45] Defronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus., 2009, 58(4): 773–795.
[46] Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Görgün CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes., 2006, 313(5790): 1137–1140.
[47] Bensellam M, Laybutt DR, Jonas JC. The molecular mechanisms of pancreatic β-cell glucotoxicity: recent findings and future research directions., 2012, 364(1–2): 1–27.
[48] Kharroubi I, Ladrière L, Cardozo AK, Dogusan Z, Cnop M, Eizirik DL. Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: role of nuclear factor-kappaB and endoplasmic reticulum stress., 2004, 145(11): 5087–5096.
[49] Cnop M, Ladriere L, Hekerman P, Ortis F, Cardozo AK, Dogusan Z, Flamez D, Boyce M, Yuan J, Eizirik DL. Selective inhibition of eukaryotic translation initiation factor 2 alpha dephosphorylation potentiates fatty acid-induced endoplasmic reticulum stress and causes pancreatic beta-cell dysfunction and apoptosis., 2007, 282(6): 3989–3997.
[50] Preston AM, Gurisik E, Bartley C, Laybutt DR, Biden TJ. Reduced endoplasmic reticulum (ER)-to-Golgi protein trafficking contributes to ER stress in lipotoxic mouse beta cells by promoting protein overload., 2009, 52(11): 2369–2373.
[51] Pétremand J, Puyal J, Chatton JY, Duprez J, Allagnat F, Frias M, James RW, Waeber G, Jonas JC, Widmann C. HDLs protect pancreatic β-cells against ER stress by restoring protein folding and trafficking., 2012, 61(5): 1100–1111.
[52] Jeffrey KD, Alejandro EU, Luciani DS, Kalynyak TB, Hu XK, Li H, Lin YL, Townsend RR, Polonsky KS, Johnson JD. Carboxypeptidase E mediates palmitate-induced beta-cell ER stress and apoptosis., 2008, 105(24): 8452–8457.
[53] Huang CJ, Lin CY, Haataja L, Gurlo T, Butler AE, Rizza RA, Butler PC. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes., 2007, 56(8): 2016–2027.
[54] Yamamoto WR, Bone RN, Sohn P, Syed F, Reissaus CA, Mosley AL, Wijeratne AB, True JD, Tong X, Kono T, Evans-Molina C. Endoplasmic reticulum stress alters ryanodine receptor function in the murine pancreatic beta cell., 2019, 294(1): 168–181.
[55] Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond., 2012, 13(2): 89–102.
[56] Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells., 2000, 6(5): 1099–1108.
[57] Hollien J, Weissman JS. Decay of endoplasmic reticulum- localized mRNAs during the unfolded protein response., 2006, 313(5783): 104-107.
[58] Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response., 2010, 40(2): 280–293.
[59] Lee AH, Chu GC, Iwakoshi NN, Glimcher LH. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands., 2005, 24(24): 4368–4380.
[60] Huang H, Yang KY, Wang RN, Han WH, Kuny S, Horn P, Zelmanovitz, Sauvé Y, Chan CB. β-Cell compensation concomitant with adaptive endoplasmic reticulum stress and β-cell neogenesis in a diet-induced type 2 diabetes model., 2019, 44(12): 1355–1366.
[61] Hwang JW, Qi L. Quality control in the endoplasmic reticulum: crosstalk between ERAD and UPR pathways., 2018, 43(8): 593–605.
[62] Xu TF, Yang L, Yan C, Wang XX, Huang P, Zhao F, Zhao LY, Zhang ML, Jia WP, Wang XD, Liu Y. The IRE1α- XBP1 pathway regulates metabolic stress-induced compensatory proliferation of pancreatic β-cells., 2014, 24(9): 1137–1140.
[63] Sharma RB, O'Donnell AC, Stamateris RE, Ha B, McCloskey KM, Reynolds PR, Arvan P, Alonso LC. Insulin demand regulates β cell number via the unfolded protein response., 2015, 125(10): 3831–3846.
[64] Burgos-Morón E, Abad-Jiménez Z, Marañón AM, Iannantuoni F, Escribano-López I, López-Domènech S, Salom C, Jover A, Mora V, Roldan I, Solá E, Rocha M, Víctor VM. Relationship between oxidative stress, ER stress, and inflammation in type 2 diabetes: the battle continues., 2019, 8(9): 1385.
[65] Bae YS, Oh H, Rhee SG, Yoo YD. Regulation of reactive oxygen species generation in cell signaling., 2011, 32(6): 491–509.
[66] Sekine N, Cirulli V, Regazzi R, Brown LJ, Gine E, Tamarit-Rodriguez J, Girotti M, Marie S, MacDonald MJ, Wollheim CB. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic beta-cells. Potential role in nutrient sensing., 1994, 269(7): 4895–4902.
[67] Yang WX, Tanaka Y, Bundo M, Hirokawa N. Antioxidant signaling involving the microtubule motor KIF12 is an intracellular target of nutrition excess in beta cells., 2014, 31(2): 202–214.
[68] Sampson SR, Bucris E, Horovitz-Fried M, Parnas A, Kahana S, Abitbol G, Chetboun M, Rosenzweig T, Brodie C, Frankel S. Insulin increases H2O2-induced pancreatic beta cell death., 2010, 15(10): 1165– 1176.
[69] Rharass T, Lemcke H, Lantow M, Kuznetsov SA, Weiss DG, Panáková D. Ca2+-mediated mitochondrial reactive oxygen species metabolism augments Wnt/β-catenin pathway activation to facilitate cell differentiation., 2014, 289(40): 27937–27951.
[70] Reczek CR, Chandel NS. ROS-dependent signal transduction., 2015, 33: 8–13.
[71] Zuo L, Zhou T, Pannell BK, Ziegler AC, Best TM. Biological and physiological role of reactive oxygen species - the good, the bad and the ugly., 2015, 214(3): 329–348.
[72] Kitamura T. The role of FOXO1 in β-cell failure and type 2 diabetes mellitus., 2013, 9(10): 615–623.
[73] Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology., 2018, 20(9): 1013–1022.
[74] Zeng C, Mulas F, Sui YH, Guan T, Miller N, Tan YL, Liu FF, Jin W, Carrano AC, Huising MO, Shirihai OS, Yeo GW, Sander M. Pseudotemporal ordering of single cells reveals metabolic control of postnatal β cell proliferation., 2017, 25(5): 1160–1175.e1111.
[75] Baumel-Alterzon S, Katz LS, Brill G, Jean-Pierre C, Li YS, Tse I, Biswal S, Garcia-Ocaña A, Scott DK. Nrf2 regulates β-cell mass by suppressing β-cell death and promoting β-cell proliferation., 2022, 71(5): 989–1011.
[76] Ehses JA, Perren A, Eppler E, Ribaux P, Pospisilik JA, Maor-Cahn R, Gueripel X, Ellingsgaard H, Schneider MK, Biollaz G, Fontana A, Reinecke M, Homo-Delarche F, Donath MY. Increased number of islet-associated macrophages in type 2 diabetes., 2007, 56(9): 2356–2370.
[77] Böni-Schnetzler M, Ehses JA, Faulenbach M, Donath MY. Insulitis in type 2 diabetes., 2008, 10(Suppl 4): 201–204.
[78] Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease., 2011, 11(2): 98–107.
[79] Cardozo AK, Ortis F, Storling J, Feng YM, Rasschaert J, Tonnesen M, Van Eylen F, Mandrup-Poulsen T, Herchuelz A, Eizirik DL. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells., 2005, 54(2): 452–461.
[80] Choudhury S, Ghosh S, Gupta P, Mukherjee S, Chattopadhyay S. Inflammation-induced ROS generation causes pancreatic cell death through modulation of Nrf2/NF-κB and SAPK/JNK pathway., 2015, 49(11): 1371–1383.
[81] Hansen JB, Tonnesen MF, Madsen AN, Hagedorn PH, Friberg J, Grunnet LG, Heller RS, Nielsen A, Størling J, Baeyens L, Anker-Kitai L, Qvortrup K, Bouwens L, Efrat S, Aalund M, Andrews NC, Billestrup N, Karlsen AE, Holst B, Pociot F, Mandrup-Poulsen T. Divalent metal transporter 1 regulates iron-mediated ROS and pancreatic β cell fate in response to cytokines., 2012, 16(4): 449–461.
[82] Yong J, Parekh VS, Reilly SM, Nayak J, Chen ZJ, Lebeaupin C, Jang I, Zhang JW, Prakash TP, Sun H, Murray S, Guo SL, Ayala JE, Satin LS, Saltiel AR, Kaufman RJ. Chop/Ddit3 depletion in β cells alleviates ER stress and corrects hepatic steatosis in mice., 2021, 13(604): eaba9796.
[83] Han J, Backa SH, Hur J, Lin YH, Gildersleeve R, Shan JX, Yuan CL, Krokowski D, Wang SY, Hatzoglou M, Kilberg MS, Sartor MA, Kaufman RJ. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death., 2013, 15(5): 481–490.
[84] Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response., 2007, 318(5852): 944–949.
[85] Yang Y, Cai ZX, Pan ZH, Liu F, Li DD, Ji YJ, Zhong JX, Luo HR, Hu SB, Song L, Yu SJ, Li T, Li JQ, Ma XH, Zhang WP, Zhou ZG, Liu F, Zhang JJ. Rheb1 promotes glucose-stimulated insulin secretion in human and mouse β-cells by upregulating GLUT expression., 2021, 123: 154863.
[86] Yang WL, Chi YJ, Meng YH, Chen ZZ, Xiang R, Yan H, Yang JC. FAM3A plays crucial roles in controlling PDX1 and insulin expressions in pancreatic beta cells., 2020, 34(3): 3915–3931.
[87] Talchai C, Xuan SH, Lin HV, Sussel L, Accili D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure., 2012, 150(6): 1223–1234.
[88] Zeeshan HM, Lee GH, Kim HR, Chae HJ. Endoplasmic reticulum stress and associated ROS., 2016, 17(3): 327.
[89] Chow J, Rahman J, Achermann JC, Dattani MT, Rahman S. Mitochondrial disease and endocrine dysfunction., 2017, 13(2): 92–104.
[90] Yoon YS, Yoon DS, Lim IK, Yoon SH, Chung HY, Rojo M, Malka F, Jou MJ, Martinou JC, Yoon G. Formation of elongated giant mitochondria in DFO-induced cellular senescence: involvement of enhanced fusion process through modulation of Fis1., 2006, 209(2): 468–480.
[91] Schultz J, Waterstradt R, Kantowski T, Rickmann A, Reinhardt F, Sharoyko V, Mulder H, Tiedge M, Baltrusch S. Precise expression of Fis1 is important for glucose responsiveness of beta cells., 2016, 230(1): 81–91.
[92] Dandona P, Thusu K, Cook S, Snyder B, Makowski J, Armstrong D, Nicotera T. Oxidative damage to DNA in diabetes mellitus., 1996, 347(8999): 444–445.
[93] Al-Aubaidy HA, Jelinek HF. Oxidative DNA damage and obesity in type 2 diabetes mellitus., 2011, 164(6): 899–904.
[94] Eguchi K, Manabe I, Oishi-Tanaka Y, Ohsugi M, Kono N, Ogata F, Yagi N, Ohto U, Kimoto M, Miyake K, Tobe K, Arai H, Kadowaki T, Nagai R. Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation., 2012, 15(4): 518–533.
[95] Ivovic A, Oprescu AI, Koulajian K, Mori Y, Eversley JA, Zhang L, Nino-Fong R, Lewis GF, Donath MY, Karin M, Wheeler MB, Ehses J, Volchuk A, Chan CB, Giacca A. IKKβ inhibition prevents fat-induced beta cell dysfunction in vitro and in vivo in rodents., 2017, 60(10): 2021–2032.
[96] Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling., 2011, 21(1): 103–115.
[97] Yang BY, Yang L, Wang YY, Maddison LA, Tang ZH, Haigh S, Gong YL, Zhang Y, Covington BA, Bosma KJ, Tong X, Page-McCaw P, Gannon M, Deng Q, Chen WB. Macrophages and neutrophils are necessary for ER stress-induced β cell loss., 2022, 40(8): 111255.
[98] Salazar J, Angarita L, Morillo V, Navarro C, Martínez MS, Chacín M, Torres W, Rajotia A, Rojas M, Cano C, Añez R, Rojas J, Bermudez V. Microbiota and diabetes mellitus: role of lipid mediators., 2020, 12(10): 3039.
[99] Schwandner R, Dziarski R, Wesche H, Rothe M, Kirschning CJ. Peptidoglycan- and lipoteichoic acid- induced cell activation is mediated by toll-like receptor 2., 1999, 274(25): 17406–17409.
[100] Gao H, Luo ZL, Ji YD, Tang KC, Jin ZM, Ly C, Sears DD, Mahata S, Ying W. Accumulation of microbial DNAs promotes to islet inflammation and β cell abnormalities in obesity in mice., 2022, 13(1): 565.
[101] Cinti F, Bouchi R, Kim-Muller JY, Ohmura Y, Sandoval PR, Masini M, Marselli L, Suleiman M, Ratner LE, Marchetti P, Accili D. Evidence of beta-cell dedifferentiation in human type 2 diabetes., 2016, 101(3): 1044–1054.
[102] Spijker HS, Ravelli RBG, Mommaas-Kienhuis AM, van Apeldoorn AA, Engelse MA, Zaldumbide A, Bonner- Weir S, Rabelink TJ, Hoeben RC, Clevers H, Mummery CL, Carlotti F, de Koning EJP. Conversion of mature human beta-cells into glucagon-producing alpha-cells., 2013, 62(7): 2471–2480.
[103] Collombat P, Mansouri A, Hecksher-Sorensen J, Serup P, Krull J, Gradwohl G, Gruss P. Opposing actions of Arx and Pax4 in endocrine pancreas development., 2003, 17(20): 2591–2603.
[104] Sosa-Pineda B, Chowdhury K, Torres M, Oliver G, Gruss P. Thegene is essential for differentiation of insulin-producing beta cells in the mammalian pancreas., 1997, 386(6623): 399-402.
[105] Ilegems E, Bryzgalova G, Correia J, Yesildag B, Berra E, Ruas JL, Pereira TS, Berggren PO. HIF-1α inhibitor PX- 478 preserves pancreatic β cell function in diabetes., 2022, 14(638): eaba9112.
[106] Preissl S, Gaulton KJ, Ren B. Characterizing cis- regulatory elements using single-cell epigenomics., 2022, doi: 10.1038/s41576-022-00509-1.
Molecular mechanism of islet β-cell functional alternations during type 2 diabetes
Chengan Lv1,2, Ruoran Wang1,2, Zhuo-Xian Meng1,2
In recent years, the incidence rate of type 2 diabetes (T2D) has risen rapidly and has become a global health crisis. Recent experimental and clinical studies have shown that islet β-cell dysfunction is an important cause of T2D and its related complications. β-cells undergo dynamic compensation and decompensation in the course of T2D. In this process, metabolic stress responses, such as ER stress, oxidative stress and inflammation, are key regulators of β-cell functional alternations. In this review, we summarize the research progress on the β-cell functional dynamics in the course of T2D, in order to deepen the understanding of the molecular mechanism of T2D, and provide reference for its precise diagnosis and clinical intervention.
β-cell;type 2 diabetes; molecular mechanism
2022-08-04;
2022-09-02;
2022-09-30
国家自然科学基金项目(编号:91857110,81722012,81670740),科技部国家重点研发计划项目(编号:2018YFA0800403,2021YFC20701903),浙江省自然科学基金项目(编号:LZ21H070001)和杭州市医学重点学科建设基金项目(No.OO20200055)资助 [Supported by the National Natural Science Foundation of China (Nos. 91857110, 81722012, 81670740), the National Key R&D Program of the Ministry of Science and Technology (Nos. 2018YFA0800403, 2021YFC20701903), the Zhejiang Provincial Natural Science Foundation of China (No. LZ21H070001), and the Construction Fund of Medical Key Disciplines of Hangzhou (No. OO20200055)]
吕承安,在读临床医学八年制本科生,专业方向:糖尿病的分子机制。E-mail: 3190100992@zju.edu.cn
孟卓贤,博士,研究员,研究方向:糖尿病的分子机制。E-mail: zxmeng@zju.edu.cn
10.16288/j.yczz.22-265
(责任编委: 周红文)
