基于CRISPR/Cas9系统制作拟南芥糖基转移酶ugt79b2/79b3双突变体纯合株系
2022-11-19郭伟岳张雨飞冀芦沙
滕 霄,郭伟岳, 李 茹,李 攀, 张雨飞, 冀芦沙
(1. 聊城大学 生命科学学院,山东 聊城 252059;2. 聊城大学 药学院,山东 聊城 252059)
CRISPR体系是在细菌中发现的一种获得性免疫系统,能抵抗外源DNA侵袭[1]。研究发现,约90%古细菌和40%细菌中含有CRISPR系统[2]。CRISPR体系是由短的高度保守的重复序列(repeats, R, 20~50 bp)及间区序列(spacers, S, 36 bp)间隔成簇排列,其5′端有前导序列(leader, L, 550 bp)和Cas蛋白基因(4~10个)[3]。研究表明,新的RS序列往往会插入到L序列及其临近的重复序列之间[4]。
Makarova等将CRISPR系统分为3种类型:I型系统由Cas1、Cas2及Cas3蛋白共同发挥基因编辑作用[5];Ⅱ型系统仅由Cas9蛋白就可以进行基因编辑[3];Ⅲ型系统中Cas6在长链pre-crRNA加工及加工后传递发挥重要作用[6]。
1 材料与方法
1.1 材料
植物材料:拟南芥(Arabidopsisthaliana)为哥伦比亚野生型(Columbia wild-type),保存于聊城大学药学院植物培养室。
菌株与载体:大肠杆菌E. coli DH5α、农杆菌GV3101均由本实验保存;克隆载体pEASY-T1 Cloning Kit、pEASY-Blunt Cloning Kit均购买于北京全式金生物技术有限公司;gRNA引物、基因序列均由上海生工生物工程有限公司合成、测序;
仪器:组织研磨仪(Tissuelyser-192)购买于上海净信生物仪器公司;
试剂与耗材:CRISPR/Cas9试剂盒购买于江苏百格生物技术有限公司;TaqDNA聚合酶、限制性内切酶、T4 DNA Ligase试剂盒、质粒提取试剂盒、DNA回收试剂盒及DNA Maker等均分子生物学耗材均购买于大连宝生物(TaKaRa)生物技术有限公司;DNA提取缓冲液购买于美国赛默飞公司;甲醇、乙醇等有机试剂均购买于北京国药集团。
1.2 gRNA引物设计
根据拟南芥信息资源数据库(TheArabidopsisInformation Resource, TAIR)查找拟南芥ugt79b2和ugt79b3的基因组序列号,分别为AT4G27560和AT4G27570,下载ugt79b2和ugt79b3全长基因序列,分别设计ugt79b2和ugt79b3的sgRNA靶点序列,见表1。
1.3 大肠杆菌DH5α感受态制备
挑取新鲜活化的大肠杆菌DH5α单菌落,将其接种到LB液体培养基中(5 mL),37 ℃,220 r/min过夜摇菌制备种子液;将种子液按照1:100的比例接种到新的LB液体培养基中(50 mL),37 ℃,220 r/min振荡培养。培养4~6 h后,紫外分光光度计测量菌液浓度,当OD600达到0.6~0.8时,停止振荡培养;将大肠杆菌培养液冰浴冷却30 min后,4 ℃,4500 r/min离心20 min,收集菌体;将细菌细胞于冰浴冷却的去离子水中重悬,4 ℃,4500 r/min离心20 min,重悬3次后,将细菌细胞悬于冰浴冷却的甘油中(10%,V/V);精确量取100 μL细菌细胞液于1.5 mL无菌离心管中,于-80 ℃超低温冰箱中保存备用。
1.4 农杆菌GV3101感受态制备
挑取新鲜活化的农杆菌GV3101单菌落,将其接种到LB液体培养基中(5 mL),28 ℃,200 r/min过夜摇菌制备种子液;将种子液按照1:100的比例接种到新的LB液体培养基中(50 mL),28 ℃,200 r/min振荡培养。培养8~10 h后,紫外分光光度计测量菌液浓度,当OD600达到0.4~0.6时,停止振荡培养;将农杆菌培养液冰浴冷却30 min后,4 ℃,4500 r/min离心20 min,收集菌体;将细菌细胞于冰浴冷却的去离子水中重悬,4 ℃,4500 r/min离心20 min,重悬3次后,将细菌细胞悬于冰浴冷却的甘油中(10%,V/V);精确量取100 μL细菌细胞液于1.5 mL无菌离心管中,于-80 ℃超低温冰箱中保存备用。
1.5 ugt79b2/79b3-Cas9载体构建
按照表1制备ugt79b2,ugt79b3 Oligo二聚体,20 μL反应体系中加入18 μL Anneal Buffer、1 μL cas-ugt79b2-F (10 μmol/L)、1 μL cas-ugt79b2-R(10 μmol/L)或18 μL Anneal Buffer、1 μL cas-ugt79b3-F (10 μmol/L)、1 μL cas-ugt79b3-R(10 μmol/L)。接着95 ℃变性5 min,然后以0.2 ℃/s缓慢降低温度进行退火,直到20 ℃。将上述制备成功的Oligo二聚体与CRISPR/Cas-9载体进行连接,具体连接体系如表2所示,25 ℃反应1 h。
1) 适当放大选择港口的中转时间差阈值条件后,由于港口选择范围的扩大使得港口群内某些核心枢纽港的泊位资源压力得到缓解,非核心枢纽港的泊位资源得到进一步利用。
反应结束后,取5.0 μL上述连接体系反应液,加入20 μL大肠杆菌感受态细胞,通过电击转化法将连接体系转化到大肠杆菌中。转化液涂布到LB平板中,37 ℃倒置培养1 d,用表1中cas9引物进行菌落PCR验证。若菌落PCR呈阳性,则摇菌扩大培养,并取部分菌液送生物公司测序。将100%测序正确阳性菌落,一部分置于-80 ℃超低温冰箱保存,另一部分提取质粒,并转入农杆菌GV3101感受态细胞中。将农杆菌转化液涂布到含有利福平(50 μg/L)和卡那霉素(50 μg/L)的LB平板中,28 ℃倒置培养2 d,用表1中cas9引物进行菌落PCR验证。若菌落PCR呈阳性,则摇菌扩大培养,一部分置于-80 ℃超低温冰箱保存,另一部分用于后续拟南芥花侵染。
1.6 PCR扩增
PCR反应体系如表3所示。
PCR程序:(1) 预热:96 ℃预变性5min;(2) PCR循环(35~40次):96 ℃变性50 s,50~60 ℃退火50 s,72 ℃延伸30 s~2 min;(3) 延伸:72 ℃延伸10 min;(4) 4 ℃保存。
1.7 拟南芥花浸染与ugt79b2/79b3-Cas9突变阳性株系筛选
将ugt79b2/79b3-Cas9GV3101菌液活化后,采用农杆菌蘸花法侵染野生型拟南芥的花序(抽薹约1 cm),侵染后,过夜黑暗处理,每隔1周侵染1次,反复侵染3次。一段时间后,收集农杆菌侵染后的拟南芥种子,并置于卡那霉素(50 μg/L)的MS平板中筛选,长势良好株系为阳性植株,移栽到土壤中继续培养。三代筛选后,获得阳性纯合株系。提取阳性纯合株系DNA,PCR鉴定阳性株系后,进一步送生物公司测序,确定是否突变。
1.8 植物基因组DNA提取
用小剪刀剪取拟南芥幼嫩叶片(1 cm3)一片,置于1.5 mL EP管中;加入直径2.0 mm的氧化锆株和100 μL DNA提取缓冲液;混合均匀后,置于组织研磨仪上震荡破碎10~60 s;4200 r/min 离心2 min,将上清液转移到新的1.5 mL EP管中,4 ℃保存,用于后续PCR扩增模板。
1.9 数据统计与分析
2 结果与分析
2.1 sgRNA靶点序列选择
从拟南芥信息资源库TAIR中下载ugt79b2和ugt79b3全长基因组序列,二者同源性高达99%,根据sgRNA软件设计ugt79b2和ugt79b3的sgRNA靶点序列,多次筛选,最终确定UGT79B2/79B3共同保守靶序列sgRNA(图1):GCCAACAAATTGGCTGAGAAAGG;利用该靶点序列设计引物,其中拟南芥ugt79b2靶点序列引物Oligo(5′-3′):cas-ugt79b2-F为CAGAAATGGGTGGTTTGAAGTTTC,cas-ugt79b2-R为AAGACTTGTAGTGACTCTTT
CCAAT;拟南芥ugt79b3靶点序列引物Oligo(5′-3′)cas-ugt79b3-F为CAGAAATGGGTGGTTTGAAGTTTC,cas-ugt79b3-R为GTTTCCTTCGATTTCTCTGGCTG。
2.2 ugt79b2/79b3-Cas9载体构建
按照上述方法构建ugt79b2/79b3-Cas9载体后,PCR扩增验证ugt79b2/79b3-Cas9阳性转化子,结果发现,ugt79b2阳性转化子的PCR条带大小为1368 bp如图2(a)所示,ugt79b3阳性转化子的PCR条带大小为1362 bp如图2(a)所示。将验证成功的阳性转化子菌液送生物公司测序,结果如图2(b)所示,测序结果与TAIR公布序列一致,100%正确,该结果表明ugt79b2/79b3-Cas9载体构建成功。
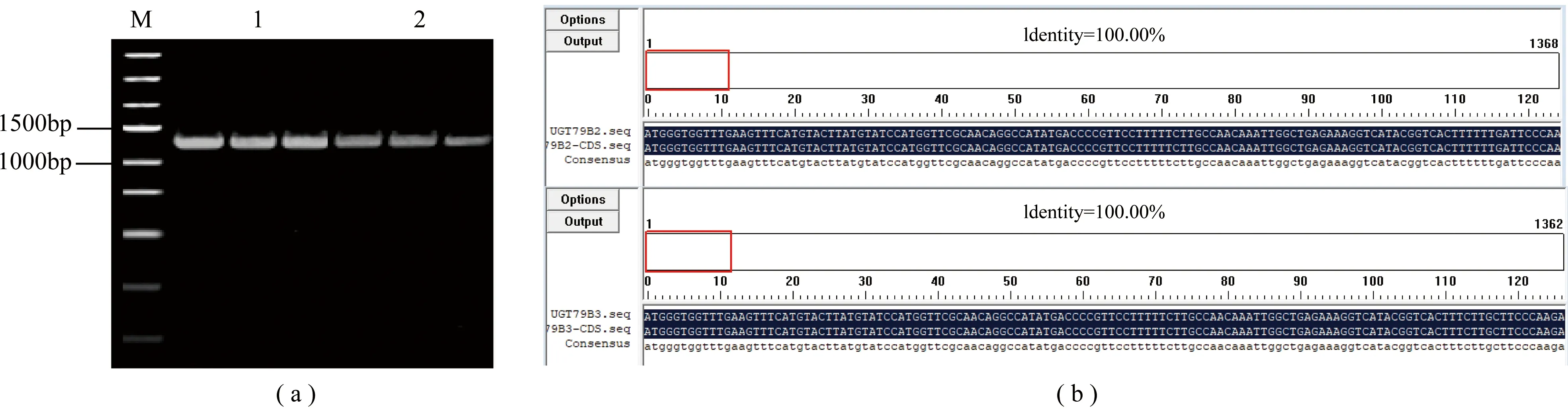
注:(a)CRISPR/ugt79b2/79b3-Cas9菌落PCR验证(M:Marker;1:UGT79B2引物验证;2:UGT79B3引物验证);(b):CRISPR/ugt79b2/79b3-Cas9菌落基因测序结果比对。图2 ugt79b2/79b3-Cas9载体构建
2.3 ugt79b2-cas9嵌合体筛选
将上述构建成功的ugt79b2/79b3-Cas9 GV3101侵染拟南芥,获得T0代种子,潮霉素筛选后,获得阳性株系99株,分别命名T1-1,T1-2,…,T1-99。提取99株幼苗DNA,以cas9-ugt79b2为引物,送公司测序,结果如表4所示,ugt79b2-cas9嵌合体筛选获得,缺A碱基7株,A碱基替换为G或C碱基12株,A碱基插入3株,缺A、G碱基2株。

表4 ugt79b2-cas9突变体植株
2.4 ugt79b2-Cas9单突变纯合体筛选
选取T1代突变株系,继续培养,获得T2代种子,潮霉素筛选后,获得阳性株系24株,分别命名T2-1-1, T2-1-2…T2-2-1, T2-2-2…T2-24-1, T2-24-2…。提取X株幼苗DNA,以cas9-ugt79b2为引物,送公司测序,结果如表5所示,ugt79b2-cas9纯合突变体选获得,缺A碱基2株,A碱基替换为G或C碱基3株,A碱基插入1株,缺A、G碱基1株。

表5 ugt79b2-cas9突变纯合体植株
2.5 ugt79b2/ugt79b3-Cas9双突变纯合体筛选
提取ugt79b2-Cas9单突变纯合体幼苗基因组DNA,以cas9-ugt79b3为引物,送公司测序,结果如图3所示,ugt79b2/ugt79b3-Cas9双突变纯合体筛选获得1株A碱基插入突变,用于后续ugt79b2、ugt79b3功能验证研究。
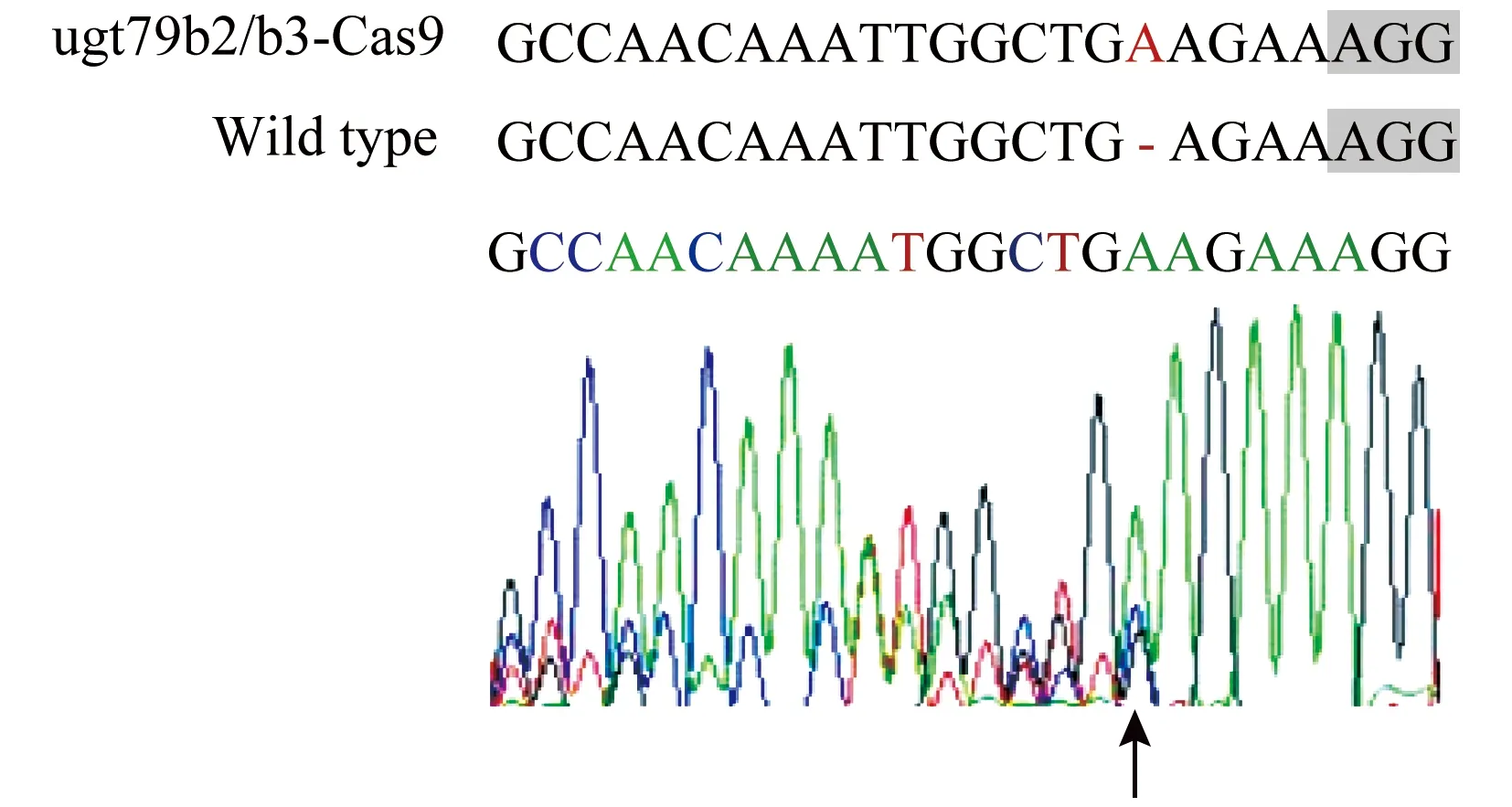
图3 ugt79b2/ugt79b3-Cas9双突变纯合体筛选
3 讨论
植物糖基转移酶是植物体内能够将各种糖基转移到小分子化合物上,如多肽类、激素类以及次级代谢产物等,改变小分子化合物特性,进而影响植物生长发育的酶类[10]。研究发现,糖基转移酶参与非生物胁迫、调节激素平衡、参与次级代谢响应、参与生物胁迫、参与解毒反应、参与信号转导等[11-16]。根据报道,糖基转移酶UGT79B2/79B3能够糖基化次级代谢物花青素类化合物,超表达拟南芥表现出抗冷、抗盐、抗旱等表型[17]。虽然拟南芥糖基转移酶UGT79B2/79B3的基因功能早已被研究,但是植物体内代谢平衡是一个非常复杂的过程,通过CRISPR/Cas9技术获得ugt79b2/ugt79b3-Cas9双突变纯合体为其他物种中ugt79b2/ugt79b3同源基因双突变体筛选提供技术支持。
CRISPR/Cas9是目前唯一优先应用于基因编辑的Ⅱ型系统。与I型和Ⅲ型系统不同,Ⅱ型系统仅需要2个RNA元件和1个Cas蛋白即可实现对DNA靶序列的切割[18]。为了进一步精简CRISPR/Cas9系统,科研人员将必需元件crRNA与tracrRNA的核心序列合并为一个sgRNA,体外实验发现,在sgRNA引导下,Cas9蛋白能够切割双链DNA[19]。随着CRISPR/Cas9试剂盒的研发,使得CRISPR/Cas9系统广泛的用于真核生物内源基因编辑,如水稻、棉花、拟南芥、小鼠、斑马鱼、果蝇等[20-25]。
CRISPR/Cas9基因编辑技术由两大核心技术组成:(1)构建sgRNA-Cas9表达载体,并将载体转化到受体细胞中发挥基因编辑作用;(2)将合成的sgRNA序列与表达纯化的Cas9蛋白导入到受体细胞中发挥切割作用[26]。链球菌(Streptococcus pyogenes)中SpCas9蛋白最先被用于基因编辑研究,该蛋白由一个HNH核酸酶结构域和一个RuvC-like结构域组成[27]。这两个结构域分别在靶DNA的PAM序列(NGG)上游3个碱基处对DNA双链进行切割,形成平末端。真核系统中,还需要在Cas9蛋白中添加一段核定位信号,以保证该蛋白能够顺利进入细胞核发挥功能。sgRNA是单链RNA,约20个碱基,可以与靶DNA序列互补配对,进而引导sgRNA-Cas9复合物在特定位点上发生切割[26]。因此,构建sgRNA-Cas9表达载体时,仅仅需要改变sgRNA中特异位点的识别序列。本研究中,根据拟南芥信息资源库TAIR中下载ugt79b2和ugt79b3全长基因组序列,二者同源性高达99%,sgRNA软件设计ugt79b2和ugt79b3的sgRNA靶点序列,多次筛选,最终确定1条sgRNA序列:GCCAACAAATTGGCTGAGAAAGG;利用该靶点序列设计引物,其中拟南芥ugt79b2靶点序列引物Oligo(5′-3′):cas-ugt79b2-F为CAGAAATGGGTGGTTTGAAGTTTC,cas-ugt79b2-R为AAGACTTGTAGTGACTCTTTCCAAT;拟南芥ugt79b3靶点序列引物Oligo(5′-3′)cas-ugt79b3-F为CAGAAATGGGTGGTTTGAAGTTTC,cas-ugt79b3-R为GTTTCCTTCGATTTCTCTGGCTG。
CRISPR/Cas9系统在植物基因组编辑应用过程中具有成本低、操作简单,且突变效率高等特点,还能够实现多个基因同时编辑,在遗传育种与基因功能研究等领域取得了高速发展。对于CRISPR/Cas9系统突变株系,科研工作者通过3种靶点分析技术对其进行分析。第1种,基于Sanger测定的靶点分析技术[28]。研究表明,基于Sanger测序的靶点分析技术比较适应于简单突变的样品。第2种,基于高通量测序的靶点分析技术[29]。当植株突变类型同时涉及简单突变、嵌合突变或者一次需要测量大量靶点突变、解析多倍体物种时,往往采用基于高通量二代测序的靶点分析技术。目前该类技术包括Cas-analyzer、CRISPR-GA、AGEseq、Hi-TOM和CRISPResso等。第3种,基于非测序手段的靶点分析技术。与上述两种方法相比,基于非测序手段的靶点分析技术可以在无法获知具体碱基变异序列时,就能够非常简单地判断该基因是否编辑成功,包括T7E1(T7 endonuclease 1)法[30]、PCR-RE(PCR/restriction enzyme)法[31]和HRM(High-resolution melting assay)法[32]等。本研究中,我们采用基于Sanger测序的靶点分析技术就能分析ugt79b2-cas9嵌合体、ugt79b2-Cas9单突变纯合体、ugt79b2/ugt79b3-Cas9双突变纯合体,测序结果显示,ugt79b2-cas9嵌合体筛选获得,缺A碱基7株,A碱基替换为G或C碱基12株,A碱基插入3株,缺A、G碱基2株;ugt79b2-cas9纯合突变体选获得,缺A碱基2株,A碱基替换为G或C碱基3株,A碱基插入1株,缺A、G碱基1株;ugt79b2/ugt79b3-Cas9双突变纯合体筛选获得1株A碱基插入突变,用于后续ugt79b2、ugt79b3功能验证研究。
