SUMO化修饰在心血管疾病中的研究进展
2022-11-16吴诚洁涂均楚李玉洁王燕丽李杨欣
吴诚洁,陆 琳,涂均楚,李玉洁,张 瑜,王燕丽,李杨欣
(苏州大学医学院附属第一医院心血管病研究所,中国江苏 苏州 215123)
心血管疾病(cardiovascular diseases,CVDs)是危害人类健康的主要疾病,目前已成为全球首位死因,且有逐年上升的趋势[1]。虽然心血管疾病的治疗策略有了很大的改善,但仍缺乏准确、有效、科学的治疗方法。目前研究认为,心血管疾病涉及多种蛋白质的差异表达及互相作用[2]。细胞内存在多种与蛋白质稳态相关的蛋白质翻译后修饰形式,主要包括:磷酸化、甲基化、糖基化、乙酰化、泛素化等,其中泛素化是介导蛋白质降解的重要方式之一,它通过将泛素(ubiquitin,Ub)结合到靶蛋白上,形成多聚泛素链,被蛋白酶体识别后引起靶蛋白降解,对维持机体的蛋白质稳态具有重要作用。目前研究已发现一些类泛素化蛋白,它们的结构与泛素分子相似,但功能却与泛素完全不同。在这些类泛素化蛋白中,小泛素相关修饰物(small ubiquitin-like modifier,SUMO)是最受瞩目的一类。SUMO分子在细胞内以化学的方式或依附或脱离其底物蛋白,对所修饰的蛋白质功能进行调控。SUMO化修饰在蛋白质与蛋白质之间的相互作用、转录调控、核质运输、DNA修复及基因组稳定性维持等方面均扮演着重要的角色,与细胞损伤、细胞内环境的异常以及某些心血管疾病的发生和发展有关。因此,SUMO化修饰可作为预防或治疗心血管疾病的潜在靶标。
1 SUMO化修饰
1.1 SUMO分子
SUMO广泛分布于真核生物细胞内,是一类大小约15 kD的高度保守小蛋白质,结构和反应方式与泛素相似,但功能不同。SUMO1分子及SUMO化修饰方式是在1996年发现的[3]。最初脊椎动物的SUMO1被命名为PIC1(PML-interacting clone 1)、UBLI(ubiquitin-like protein 1)、Sentrin[4],其能在不基于特定底物的情况下,调控底物蛋白的功能。真核细胞存在4种SUMO蛋白(SUMO1/2/3/4)和6种SUMO特异性蛋白酶(Sentrin/SUMO-specific protease,SENP)——SENP1/2/3/5/6/7[5](表1)。SUMO蛋白主要分布在细胞核,其中SUMO1在核膜、核仁、核质都有分布,而SUMO2/3则主要分布于核质。SUMO2和SUMO3仅相差3个氨基酸,两者与SUMO1的同源性约为47%。由于SUMO2和SUMO3很相似,因此一般把二者称为SUMO2/3。SUMO4是由人TAB2(TGF-beta activated kinase 1 binding protein 2)基因的一个内含子所编码,主要在肾、淋巴结和脾脏中表达。SUMO化修饰靶蛋白的方式有3种类型:靶蛋白单个位点的单SUMO化修饰、靶蛋白多个位点的单SUMO化修饰和靶蛋白的多聚SUMO化修饰(图1)。SUMO2/3可以使其底物蛋白发生多聚SUMO化,而SUMO1可以使其底物蛋白单SUMO化或充当聚SUMO2/3链的终止子。SUMO特异性蛋白酶,也称去SUMO化蛋白酶,在SUMO化修饰循环通路中起着关键作用,一是催化SUMO分子由前体变成活性形式,二是切断SUMO分子与靶蛋白之间形成的异肽键,实现去SUMO化。
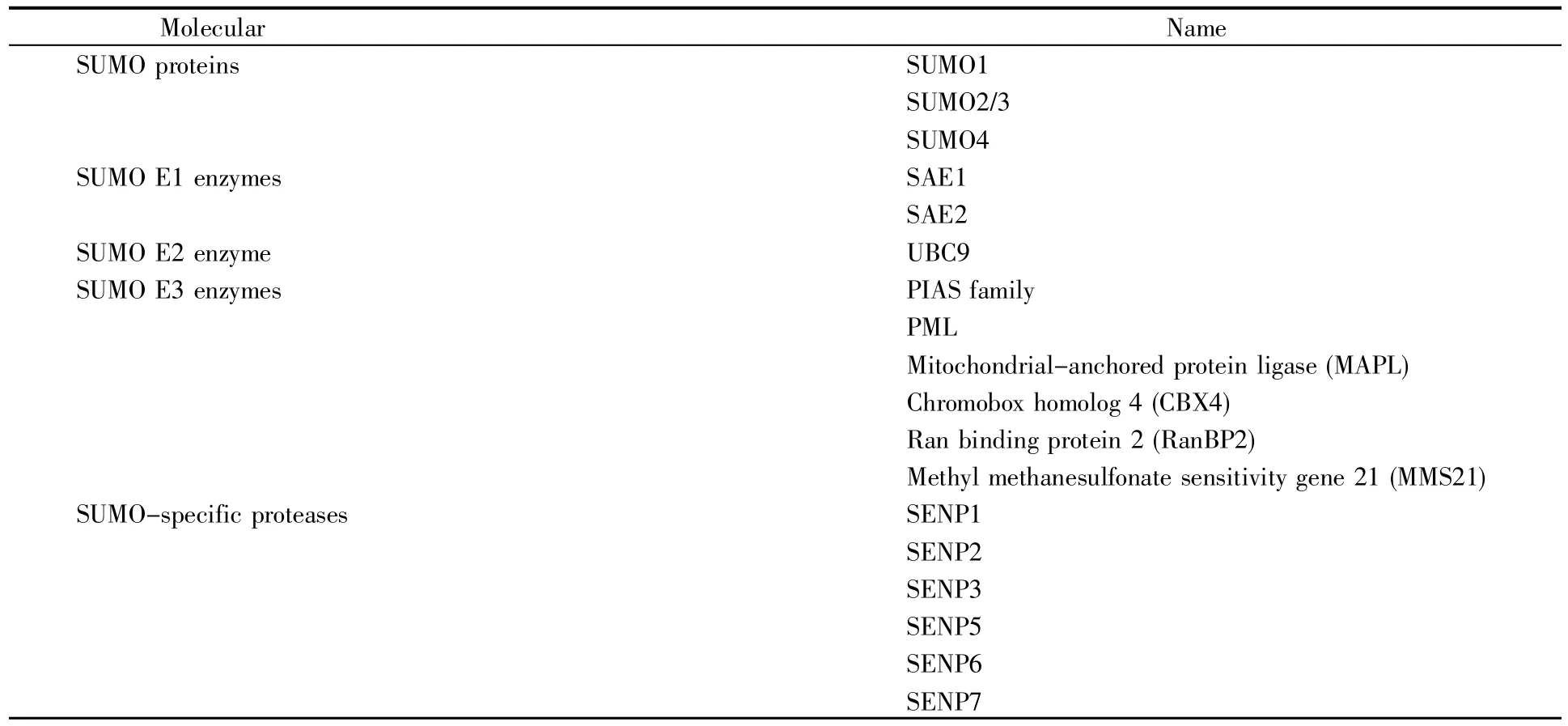
表1 SUMO化修饰过程中的关键分子Table 1 Key molecules in the process of SUMOylation
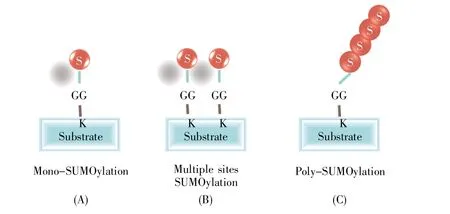
图1 多种靶蛋白的SUMO化修饰方式S:SUMO分子;GG:成熟SUMO分子C端暴露的甘氨酸残基;K:底物蛋白上的赖氨酸残基。Fig.1 SUMOylation of multiple target proteinsS:SUMO molecule;GG:The exposed glycine residue at the C-terminus of a mature SUMO molecule;K:The lysine residue on the substrate protein.
1.2 SUMO化修饰过程
SUMO修饰是一种重要的蛋白质翻译后修饰形式,可以调控底物蛋白的多种功能以及细胞中的许多应激反应。其因修饰过程与泛素化相似,也被称作类泛素化修饰。在经典的SUMO化修饰中,大多数底物蛋白都存在一段共识序列ΨKXE。其中,Ψ代表疏水性氨基酸,如异亮氨酸、甲硫氨酸、苯丙氨酸等;K代表赖氨酸,是SUMO的结合位点;X代表任意氨基酸;E代表谷氨酸[6]。SUMO化包括3个反应步骤:激活、转移、连接。SUMO激活酶、转移酶和连接酶(分别为E1、E2和E3)在SUMO化的3个反应步骤中依次起作用(图2)。激活:SUMO分子以前体非活性形式存在于细胞核中,通过SENP分子的内肽酶活性将其裂解后成为成熟的SUMO分子进入级联反应。SUMO分子在ATP的作用下与E1激活酶[SAE1(SUMO1 activating enzyme subunit 1)和SAE2组合形成的复合物]结合而活化,该反应在SUMO分子的C端与E1激活酶之间形成高能硫酯键时结束。转移:激活后,SUMO特异性结合酶UBC9(ubiquitin-like protein SUMO1 conjugating enzyme;唯一的E2转移酶)与SAE2相互作用,并和E1-SUMO分子中间体结合。SUMO分子从E1复合体转移到UBC9活性位点的半胱氨酸残基上。连接:UBC9催化成熟的SUMO分子与底物蛋白的赖氨酸残基形成异肽键,从而完成结合。E3数量最多,最常见的为PIAS(protein inhibitor of activated STAT)家族蛋白。在SUMO化修饰过程中,E3一般只是增加SUMO化修饰的效率,并不起决定性作用,在没有E3的情况下,该过程一样可以发生。与泛素化类似,SUMO化修饰也是一个动态可逆的过程。SENP酶可以切断靶蛋白与SUMO分子形成的异肽键,实现去SUMO化。SUMO化与去SUMO化的动态平衡对疾病的发生发展至关重要。
1.3 SUMO化与泛素化
蛋白质SUMO化和泛素化修饰的调节机制,以及二者之间的相互关系具有精细性和复杂性[7]。SUMO化和泛素化是蛋白质翻译后修饰的重要方式,都能够调节蛋白质功能,参与细胞周期调控、基因转录活性和信号转导等细胞活动。泛素分子通常使蛋白质底物发生多聚泛素化后经26S蛋白酶体,即泛素-蛋白酶体系统(ubiquitin-proteasome system,UPS)降解。SUMO是一种类泛素分子,主要调节蛋白质的相互作用、细胞内定位和活性等。当SUMO分子和泛素分子修饰同一底物时,SUMO分子由于与泛素竞争靶蛋白上的赖氨酸位点,所以可能会阻止底物被泛素化降解[8],因而两种修饰方式存在一种拮抗关系。但有研究发现二者也存在协同关系[9],机体内存在一种SUMO依赖的泛素连接酶——RNF4蛋白家族,某些底物的SUMO化能够激活该酶,启动UPS降解底物[10]。在急性早幼粒细胞白血病(acute promyelocytic leukemia,APL)中,三氧化二砷诱导早幼粒细胞白血病蛋白(promyelocytic leukemia protein,PML)-维甲酸受体(retinoic acid receptor alpha,RARA)致癌基因编码的融合蛋白降解,导致白血病细胞分化并产生临床缓解[11]。研究发现,PML是RNF4在体内的一个底物。当三氧化二砷治疗后,RNF4识别PML的SUMO2/3多聚泛素链,介导PML的泛素化降解,阻止PML在核中的积累,从而起到治疗APL的作用[12]。因此,泛素化和SUMO化在机体内协同与竞争关系的平衡决定了生物体各项生命活动的正常进行。
1.4 去SUMO化修饰
SUMO化修饰是一个动态可逆的过程,蛋白质发生SUMO化修饰的同时,去SUMO化修饰也会随之发生。去SUMO化修饰由SUMO特异性蛋白酶维持,主要是SENP家族,包括:SENP1、SENP2、SENP3、SENP5、SENP6和SENP7六种。SUMO化和去SUMO化过程的失控可能会打破细胞内的动态平衡,进而推动疾病的发生发展。二者之一发生异常都会导致动态紊乱,造成疾病。
2 SUMO化修饰的功能
SUMO化修饰已被证明能通过多种途径影响生命进程。一方面,SUMO化修饰通过影响蛋白质稳定性及定位、细胞周期、DNA修复、转录调控等过程维持细胞正常功能[13~15];另一方面,SUMO化修饰也会对细胞造成损伤,从而促进疾病的发生发展[16~17]。此外,SUMO化修饰还可以调节细胞骨架、线粒体功能、离子通道活性等[18~19]。
2.1 调节细胞命运
由于SUMO化的靶蛋白众多,所以这种修饰发挥功能的方式是多样的[20]。SUMO化修饰与泛素化修饰结合靶蛋白上相同的氨基酸,故二者可能存在竞争关系[21]。SUMO化修饰竞争泛素化修饰,抵抗蛋白酶体的降解,从而增加蛋白质的稳定性。已有研究表明,肌浆网/内质网钙ATP酶2a(sarcoplasmic/endoplasmic reticulum Ca2+ATPase 2a,SERCA2a)在K480和K585位点可以与SUMO1分子结合并发生SUMO化,从而影响其自身稳定性,并发挥钙离子再摄取的功能,促进心脏收缩,改善心衰表型[22]。SUMO分子与靶蛋白的结合也会影响蛋白质在细胞中的定位。研究表明,调节性T细胞中的转录调节蛋白BACH2(BTB domain and CNC homolog 2)可以发生SUMO化,而SENP3介导的去SUMO化可抑制BACH2的核输出,从而抑制与CD4 T效应细胞分化相关的基因[23]。SUMO化通过影响细胞周期的进程在维持机体的稳态中发挥着重要的作用。在人类癌症中,胞外信号调节激酶(extracellular signal-regulated protein kinase,ERK)、丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)级联介导的有丝分裂信号被Ras癌基因过度激活。致癌基因Ras通过抑制MEK(MAPK-ERK激酶)的SUMO化能有效激活ERK通路,促进细胞分化、增殖和恶性转化,从而诱导癌变[24]。在DNA损伤中,SUMO化修饰也扮演着不可或缺的角色。CtIP(CtBP interacting protein)蛋白可促进DNA末端切除,在同源重组早期发挥作用,可以使受干扰的复制不被过度的核降解。已有研究表明,CtIP蛋白可以被SUMO2分子修饰,并且在SUMO化整体受到抑制时,CtIP无法被招募到DNA损伤的位点,从而造成DNA损伤加剧[25]。此外,SUMO化也可以通过转录调控来调节细胞进程。研究者利用SLAM-seq和ChIP-seq研究了脂肪细胞分化过程中,SUMO化途径对新生基因转录的调控,发现SUMO化途径在脂肪细胞分化中具有双重功能[26]。SUMO化修饰促进前脂肪细胞特异性基因的初始下调,同时促进成熟脂肪细胞转录程序的建立。另有研究发现,PPARγ/RXR(peroxisome proliferator-activated receptor γ/retinoid X receptor)等特定转录因子的SUMO化及其辅因子与成脂基因的转录相关[27]。综上可知,SUMO化修饰在调节细胞命运方面有着重要的作用。
2.2 对细胞造成损伤
与泛素化类似,SUMO化修饰与去SUMO化修饰是一个动态平衡的过程。若该平衡被破坏,SUMO化通路也会通过不同的方式对细胞造成损伤[28]。
目前已有研究证明,SUMO化通路的异常可以通过影响氧化应激对细胞造成损伤。SENP7蛋白具有感知氧化应激、维持CD8+T细胞的代谢状态和抗肿瘤的功能。SENP7缺陷的CD8+T细胞表现出糖酵解和氧化磷酸化减少,导致体外增殖减弱,体内抗肿瘤功能减弱;CD8+T细胞衍生的活性氧(reactive oxygen species,ROS)触发胞浆SENP7介导的磷酸酶和紧张素同系物(phosphatase and tensin homolog,PTEN)蛋白的去SUMO化,从而促进PTEN降解并防止PTEN依赖的代谢缺陷[29]。SUMO化也可以通过热休克对细胞造成损伤。Kaiso是BTB/POZ锌指家族成员,参与肿瘤进展、细胞周期控制、凋亡和Wnt信号转导。在不同的启动子环境中,它可以作为转录抑制因子或激活因子发挥作用。研究发现,Kaiso蛋白可能通过自身的SUMO化调控热休克过程[30]。SUMO化的Kaiso可以激活转录,而Kaiso的去SUMO化形式则保持了Kaiso作为阻遏子的能力。正常生理条件下,肾源细胞系中的Kaiso蛋白在K42位点发生单SUMO化,进而发挥转录激活因子的功能。在细胞发生热休克后,Kaiso快速去SUMO化,激活本身阻遏子的功能,这导致了离子转运、血压和免疫应答相关基因的失调[30]。此外,有研究表明,来自RNA和DNA病毒家族的蛋白质都可以通过SUMO偶联修饰,促进病毒复制,而病毒可以通过与SUMO途径的相互作用来控制SUMO修饰的整个过程[31]。
2.3 其他作用
SUMO化除了可以调节细胞命运、对细胞造成损伤外,还有一些其他的作用。例如,SUMO化与细胞骨架功能有关。波形蛋白(vimentin,VIM)是参与细胞骨架组织和细胞运动的Ⅲ型中间丝蛋白,在K439和K445位点被SUMO化。VIM的SUMO化是其动态分解的必要条件,表达非SUMO化VIM突变体的细胞迁移水平降低[32]。SUMO化可以影响线粒体功能。研究表明,在肝缺血再灌注损伤(ischemia-reperfusion injury,I/R injury)中,动力相关蛋白1(dynamin-related peptide 1,Drp1)的磷酸化可以防止线粒体裂变,从而保护肝脏;Drp1的活性受到SUMO化修饰的调控,影响线粒体裂变的过程;肝脏再生增强剂(augmenter of liver regeneration,ALR)显著降低Drp1的SUMO化,减弱I/R损伤诱导的线粒体裂变,保持线粒体的稳定性和功能,从而使肝细胞免受I/R损伤诱导的凋亡[33]。此外,SUMO途径已被证明可以控制离子通道功能。在人类心脏中,NaV1.5是一种可以通过INa(一种快速激活和失活的Na+电流)的电压门控钠离子通道,决定了动作电位的上升和持续时间。有研究报道,在心肌缺血患者中,心脏中钠电流升高和动作电位延长,并通过Nav1的SUMO化引起心律失常[34]。K2P1是一个K+选择性的、pH敏感的、由可逆肽链调控的开放整流通道。SUMO偶联酶存在于质膜中,与K2P1结合,并修饰K2P1的K274位,从而诱导K2P1在质膜上的表达,而SENP1介导的去SUMO化又能抑制其表达。因此,K2P1的活性可以通过SUMO化修饰和去SUMO化修饰被严格调控[35]。
3 SUMO化修饰通路与心血管疾病
心血管发育是一个复杂的过程,需要各种细胞活动、转录因子和信号转导途径之间的高度协作,而在其中发挥作用的蛋白质又受许多蛋白质翻译后修饰调控。蛋白质翻译后修饰包括磷酸化、乙酰化、S-亚硝基化、糖基化和泛素化等。目前,磷酸化与泛素化已被证明在心血管疾病中有不可或缺的作用。泛素化修饰对蛋白质稳态具有调控作用,而蛋白质稳态又与心血管疾病息息相关。SUMO化作为一种类泛素化修饰也与蛋白质的稳态相互关联。近期研究也揭示,SUMO化修饰通路正在成为心血管疾病中的重要参与者[15,36~37](表2)。

表2 底物SUMO化在心血管疾病中的作用Table 2 The roles of substrate SUMOylation in CVDs
3.1 SUMO化与心力衰竭
心力衰竭(heart failure,HF)简称心衰,是指由于心脏的收缩和舒张功能发生障碍,不能将静脉回心血量充分排出心脏,导致静脉系统血液淤积,动脉系统血液灌注不足,从而引起心脏循环障碍[38]。心衰不是一个独立的疾病,而是心脏疾病发展的终末阶段[39]。
研究显示,在实验动物和人类心衰心脏中,内源性SUMO1表达降低,导致SUMO1修饰的SERCA2a减少[22]。SERCA2a是心肌细胞兴奋-收缩偶联过程中钙重新摄取到肌浆网中的关键ATP酶。SERCA2a在K480和K585位点的SUMO1修饰增加了其稳定性和ATP酶活性[40]。腺病毒介导的SUMO1递送恢复了心衰动物模型中的SERCA2表达和活性。重要的是,SUMO1基因递送改善了动物模型中的心脏射血分数[41]。敲除SERCA2a导致体外和体内心脏功能严重受损,且SUMO1过表达也无法恢复。因此,SUMO化作为一种重要的翻译后修饰形式,可以调节SERCA2a的功能并为心衰新治疗策略的设计提供新思路[22]。SUMO1在心脏中的重要性也由负调节SUMO1表达的miR-146a证实。在动物和人类发生心衰时,miR-146a的表达增加,并且miR-146a的过表达会降低体内SUMO1、SERCA2a的表达和心脏收缩性;研究进一步表明,miR-146a通过心衰心脏中的成纤维细胞分泌的细胞外囊泡递送至心肌细胞,以减少SUMO1的表达[42]。
另外,SUMO化修饰与衰老指标P21存在一定的关联。REGγ是一种蛋白酶体激活因子,能特异性结合并活化20S蛋白酶体,以ATP和泛素非依赖途径降解目的蛋白质。研究证明,REGγ能介导蛋白酶体对P21的降解[43]。Wu等[44]发现,REGγ在体内和体外都可以与SUMO1结合发生SUMO化。SUMO偶联对REGγ进行翻译后修饰,介导了REGγ的胞质易位,并增加了这种蛋白酶体激活剂的稳定性。由于REGγ的SUMO化缺陷突变体对P21的亲和力降低,因此P21的降解减少,蓄积增加,促进了衰老的发生[44]。除了SUMO1对心脏衰老有影响,还有研究发现SUMO2/3修饰蛋白在终末期心衰患者的心脏样本中也增加[45]。去SUMO化酶SENP5的过表达也会导致心肌病,在SENP5转基因小鼠的心脏中,Drp1维持低SUMO化状态导致线粒体功能失调[46]。这些研究表明SUMO化修饰对心衰具有调控作用,暗示了SUMO化修饰对心衰患者的治疗潜力。
3.2 SUMO化与心肌梗死
急性心肌梗死是冠状动脉急性、持续性缺血缺氧所引起的心肌坏死[47]。心肌I/R损伤是造成心肌梗死患者预后不良的关键因素之一[48]。
已有研究发现,SUMO化相关酶参与了心肌I/R过程,影响了疾病的进程。去SUMO化酶SENP1可以修复心肌I/R损伤。在人、小鼠以及大鼠心肌I/R后,SENP1水平均升高。在小鼠心肌I/R损伤后,SENP1敲除小鼠的收缩功能降低,心肌梗死面积增大,促使心脏功能恶化。SENP1调控缺氧诱导因子1α (hypoxia inducible factor 1α,HIF1α)的表达,这是心肌I/R过程中一个关键的保护因子。HIF1α的过表达逆转了SENP1敲除对细胞死亡的恶化作用[49]。
在心肌梗死动物模型中,ERK5被SUMO2/3高度修饰[50]。在暴露于H2O2或高葡萄糖(两种众所周知的糖尿病介质)的心肌细胞中,人们也可观察到ERK5的SUMO化。SUMO化的ERK5转录活性降低,这可能是心肌梗死后糖尿病患者心衰恶化的原因[51]。通过与丝裂原活化蛋白激酶激酶5(MAP kinase kinase 5,MEK5)结合,ERK5 的SUMO化减弱,而MEK5a过表达的转基因小鼠在心肌梗死后ERK5的SUMO化也降低。因此,ERK5的SUMO化是应激诱导的糖尿病性心肌病发展的关键因素。SUMO2/3特异性蛋白酶SENP3是缺血梗死后细胞存活的重要因素。在I/R模型中,研究人员检测了心肌内SUMO化靶蛋白和SENP3的水平变化,发现心肌缺血后SENP3丢失90%,I/R后丢失80%,并且shRNA(short hairpin RNA)介导的SENP3基因敲除导致再灌注时细胞死亡率增加,说明心脏缺血极大地改变了SENP3的水平,这可能是心脏I/R后细胞死亡的机制之一[52]。
3.3 SUMO化与心肌病
心肌病是一组异质性心肌疾病,由不同病因引起心脏机械和电活动的异常,表现为心室不适当的肥厚或扩张。严重心肌病会引起心血管性死亡或进展性心衰[53~55]。心肌素(myocardin)属于 SAP(SAF-A/B,Acinus,PIAS)结构域家族,仅在胚胎的心肌和平滑肌发育过程中表达,并在心肌细胞肥大中起关键作用。心肌素通过调节心肌细胞的稳定性和代谢活性影响心肌细胞的生长,通过血清效应因子(serum response factor,SRF)的共激活因子促进平滑肌细胞分化。心肌素的异常表达会导致肥厚型心肌病,其突变可能造成心血管发育异常[56]。据报道,心肌素在K445位点被SUMO1 SUMO化,SUMO1和E3 PIAS1对心肌素的SUMO化逆转了心肌素基因的表达,加重了心肌肥厚,表明心肌素的SUMO化在心脏肥大发展中起着重要作用[57]。线粒体生物发生和心脏能量代谢的缺陷是导致心肌病的关键因素。SENP1可以通过调节过氧化物酶体增殖物激活受体γ共激活因子1α (peroxisome proliferator-activated receptor γ coactivator 1α,PGC1α)的转录活性,调控线粒体生物发生和线粒体功能[58]。有研究证明,心肌病的发病机制与SENP1介导的线粒体异常调节有关,病变心脏中SENP1的上调是通过钙调神经磷酸酶-NFAT/MEF2C-PGC1α 通路介导的[59]。
3.4 SUMO化与动脉粥样硬化
动脉粥样硬化是慢性炎症性疾病,涉及内皮激活、内皮功能障碍和局部炎症反应等多个病理步骤。近期研究发现,SUMO化在动脉粥样硬化发生发展中起到重要的调控作用[60]。
有报道称,血流紊乱可诱导内皮细胞的促炎和凋亡反应,导致内皮细胞功能失调,进而导致动脉粥样硬化[61]。虽然已有研究证明,在内皮细胞凋亡和炎症过程中能检测到P53和ERK5的SUMO化,但在促动脉粥样硬化流动条件下,其机制在很大程度上仍是未知的[62]。在血流干扰条件下,P53和ERK5的SUMO化有助于动脉粥样硬化斑块的形成,参与这一信号转导的分子将成为控制内皮细胞功能障碍和动脉粥样硬化形成的关键靶点[63]。PIAS1是一种SUMO E3连接酶,同时也是一种转录阻遏因子。MAPK活化的蛋白激酶2是一种促炎激酶,能磷酸化PIAS1的Ser522残基,也可以发挥独特的抗炎作用。蛋白激酶2的激活增强了P53的SUMO化,但是PIAS1磷酸化突变体PIAS S522A却降低了P53的SUMO化,这表明PIAS1 S522磷酸化在其SUMO连接酶活性中起着关键作用。MAPK激活的蛋白激酶2通过增强PIAS1 S522的磷酸化介导PIAS1的转抑制和增加SUMO连接酶活性,从而限制内皮炎症[64]。除了炎症反应,巨噬细胞对动脉粥样硬化病理发生过程中的脂质代谢也具有重要的调节作用。肥胖和代谢综合征日益被认为是心血管疾病的主要危险因素。Krüppel-like转录因子 5(Krüppel-like transcription factor 5,KLF5)是一个重要的能量代谢调节因子。已有研究证明,KLF5的SUMO化在涉及PPARδ的脂质代谢转录程序中起分子开关的作用[65]。
3.5 去SUMO化酶SENP在心血管疾病中的作用
SENP是去SUMO化酶,其家族成员的作用各不相同。SENP1是一种核质穿梭蛋白质,能催化SUMO1、SUMO2/3修饰的靶蛋白发生去SUMO化。SENP2与SENP1相似,也能够对SUMO1、SUMO2/3起作用,但是二者的底物不同。比如:SENP2可以调控磷脂酶Cβ4(phospholipase Cβ4,PLCβ4)的核运输,起到维持细胞稳态的作用,而SENP1则不能[66]。在机体中,促炎因子的存在导致SENP1升高,造成下游靶蛋白去SUMO化升高,进而改变下游靶蛋白功能,影响疾病的进程[67]。在人类肥厚和衰竭心脏中,研究人员观察到SENP1的表达增加,并证明SENP1可以保护心肌细胞,使其免受肥厚性生长刺激[59]。相反,心脏特异性过表达SENP2却导致心肌病和心脏功能障碍,引起冠心病房间隔缺损小鼠过早死亡。免疫生化结果显示,与野生型相比,SENP2过表达小鼠心脏的心肌细胞增殖减少;存活的SENP2过表达小鼠生长发育迟缓,随着年龄的增长出现心肌病,心功能受损[68]。
SENP3和SENP5都是核仁蛋白质且具有很高的序列同源性,二者主要特异性结合SUMO2/3。目前SENP3在心脏中的作用颇具争议。SENP3在小鼠心脏中表达上调取决于ROS的产生,以此应对心脏的I/R损伤。SENP3敲低可以显著减少I/R损伤诱导的梗死面积并改善心功能。在心脏中,SENP3主要通过抑制内质网应激和线粒体介导的凋亡途径改善心肌细胞凋亡。SENP3的过表达显著加重了心脏的I/R损伤[69]。这些研究显示SENP3对心脏具有损害作用。相反,也有研究证明了SENP3对心肌细胞具有保护作用[70]。在缺血和再灌注期间的不同研究中,SENP3在心脏中的表达水平变化很大,这可能与SENP3在I/R后的细胞内定位发生了改变有关。有研究证明,在缺血期间SENP3的细胞质部分显著减少,同时核部分显著增加,表明缺血后SENP3可能重新定位于细胞核[52]。目前,研究者已在人的衰竭心脏中观察到SENP5的表达增加,并且发现在小鼠心脏中过表达SENP5会导致细胞增殖受损以及细胞凋亡增多,从而导致心肌病[46]。SENP5的心脏特异性过表达与线粒体裂变有关,而Drp1的寡聚化是线粒体裂变的重要因素。有研究报道,心肌特异性过表达SENP5会降低SUMO2/3缀合的Drp1水平,并诱导细胞凋亡,最终导致成年小鼠心肌病的发生[71]。
SENP7在细胞质中表达的比例较小,其和SENP6主要存在于核质中。二者主要切割多聚SUMO2/3链,从而起到拮抗SUMO化修饰的作用[72~73]。SENP6 和 SENP7 在去除 PML 的 SUMO 化及核体(nuclear bodies,NBs)形成过程中起关键作用,从而调节NBs动力学和多种细胞功能[74]。三氧化二砷注射液通过诱导PML的SUMO化和NBs形成上调TGF-β1,从而增加心肌纤维化;SUMO E2缀合物UBC9被沉默后,PML的SUMO化受到抑制,从而减少横向主动脉缩窄小鼠中心脏纤维化的发展[75]。这些结果均证明,SENP家族介导的去SUMO化修饰在心脏中发挥着至关重要的作用。
4 SUMO化修饰与外泌体在心血管疾病中的研究
细胞外囊泡(extracellular vesicles,EVs)是一种由细胞释放到细胞外基质的膜性小囊泡,参与细胞通信、细胞迁移、血管新生和肿瘤细胞生长等过程,广泛地存在于各种体液和细胞上清中,并且稳定携带一些重要的信号分子[76]。根据产生方式、体积和分子标志物的不同,EVs可分成3类,即外泌体(exosome,Exo)、细胞衍生微粒(cell-derived microparticle)和凋亡小体(apoptotic body)[77]。外泌体是EVs中体积最小的一类,直径30~100 nm。细胞质中的内体膜先向内体腔内凹陷,形成多囊内体,同时募集细胞质中的各种蛋白质、核酸、脂质进入多囊内体的各个小囊泡中,随后多囊内体迁移到细胞膜的胞质面,与细胞膜发生膜融合,释放出内部的小囊泡,即外泌体[78]。
近年来的研究表明,外泌体能够参与心血管疾病的调控。例如:心肌成纤维细胞分泌的外泌体将miR-133a输送至心肌细胞,心肌细胞中升高的miR-133a靶向果蝇ELAV样1蛋白[(embryonic lethal,abnormal vision,Drosophila)-like 1],抑制焦亡的发生,从而治疗I/R损伤[79]。间充质干细胞来源的外泌体通过递送circ-0001273,抑制心肌梗死后心肌细胞凋亡的发生,进而改善心肌梗死后的心脏功能[80]。以上研究提示,外泌体调控心血管疾病进程的主要分子机制是递送其中包含的众多非编码 RNA,如 miRNA、lncRNA、circRNA等,这也表明外泌体的生成过程中可能存在特异的RNA分选机制。目前有研究表明,SUMO化修饰与外泌体中成分的分选机制有关。外泌体的生成有依赖于内吞体分选转运复合体(endosomal sorting complex required for transport,ESCRT)和不依赖于ESCRT的途径[81]。依赖于ESCRT的外泌体生成过程不仅有泛素化修饰的参与,也有SUMOylation、NEDDylation和 ISGylation等修饰的参与[82]。SUMO化修饰在外泌体的生物发生中起重要作用,外泌体富含的miRNA的特异序列(GGAG),已被鉴定为Exo-motif。Exo-motif可被人核内不均一核糖核蛋白A2B1(human heterogeneous nuclear ribonucleoprotein A2B1,hnRNPA2B1)和hn-RNPA1特异性识别,从而调控这些miRNA选择性进入外泌体[83]。有研究报道,hnRNPA2B1在外泌体中大部分被SUMO化后,会识别特定的miRNA并将其包装到外泌体中,继而使miRNA运输到靶细胞中发挥作用[84];SUMO化修饰在自噬相关基因5(autophagy-related gene 5,ATG5)的协助下将α-突触核蛋白包埋到外泌体中,这表明SUMO化修饰是特定分子被分类到外泌体中的重要调节剂[85]。另外,已有研究证明SUMO化修饰通过EVs的运输作用调控心衰后的心脏功能。在心衰后,成纤维细胞可以分泌一种含有miR-146a的EV,其将miR-146a传递到心肌细胞,心肌细胞中的miR-146a可介导SUMO1分子的下调,从而导致SERCA2a蛋白的稳定性下降,最终造成心脏收缩功能障碍[42]。这启示,我们可以利用外泌体的“货物载体”功能调控SUMO化修饰,进而实现心血管疾病的靶向治疗。虽然SUMO化修饰可以在外泌体生物发生以及外泌体的运输过程中发挥重要作用,但是SUMO化修饰是否能够通过介导外泌体的生物发生以及基于外泌体的递送作用来调控心血管疾病,仍需要继续深入探究。随着研究的深入,我们相信在SUMO化修饰与外泌体共同靶向调控心血管疾病方面将会取得更多令人鼓舞的进展。
5 总结与展望
蛋白质翻译后修饰是重要的生物功能调控机制,其重要性不亚于转录和蛋白质表达调控,并且其复杂性更甚。即使是几种常规的蛋白质翻译后修饰,目前的研究也只是揭其冰山一角,还有很多的功能以及修饰种类有待进一步的探索。SUMO化修饰是继泛素化、磷酸化、乙酰化、甲基化和糖基化等常规修饰的又一种重要的蛋白质翻译后修饰形式。SUMO化修饰的过程与泛素化类似,但该修饰发挥的生物学功能又与泛素化大不相同,是近几年蛋白质翻译后修饰的研究热点。目前,SUMO化修饰在心血管疾病方面的研究虽有很多,但针对不同类型的疾病,包括心衰、心肌梗死、缺血性心肌病和动脉粥样硬化等,其作用及机制依旧不明确。SUMO化如何通过影响蛋白质稳定性及定位、细胞周期、DNA修复、转录调控等过程维持细胞正常功能;如何通过调节细胞骨架、线粒体功能、离子通道活性等过程影响疾病的发生发展,都需要进一步研究。
虽然心血管疾病的治疗策略有了很大的改善,但目前仍缺乏准确、有效、科学的治疗。靶向治疗是未来心血管疾病治疗的有效方式之一。外泌体具有货物运输功能,在靶向治疗心血管疾病方面具有良好的临床应用前景,因此,探究是否能够利用外泌体调控细胞中的SUMO化修饰,来达到靶向治疗心血管疾病的作用,将有助于更好地了解SUMO化在心血管疾病发生发展中的作用,并研发靶向治疗心血管疾病的精准策略。
另外,以往的蛋白质组学研究发现,多种不同类型的蛋白质翻译后修饰,如磷酸化、泛素化、甲基化、SUMO化等,存在复杂的交互作用,进而共同影响疾病的发生[86]。SUMO化不仅能调节靶蛋白的功能和定位,还可以影响其他翻译后修饰的过程,而且可能与其他翻译后修饰共同调节相同的生理病理过程。因此,在临床治疗中考虑SUMO化和其他翻译后修饰对心血管疾病进展的协同作用,探索靶蛋白SUMO化与其他蛋白质翻译后修饰之间的动态平衡,将是未来需要深入研究的方向。将SUMO化与其他蛋白质翻译后修饰形式之间复杂的关系作为心血管疾病治疗研究的切入点,可能会为心血管疾病治疗打开一扇全新的大门。
利益声明:所有作者均声明不存在利益冲突。
