阿尔茨海默病的Aβ级联机制研究进展
2022-11-16李明成米彩云王虎平
李明成,周 君,米彩云,王虎平
(甘肃中医药大学,中国甘肃 兰州 730000)
阿尔茨海默病(Alzheimer’s disease,AD)是一种中枢神经系统退行性疾病,其病程长、发展呈渐进性且不可逆,早期仅表现为短期记忆能力的受损,而长期记忆尚保存完好,随着病情进展到中、晚期,患者常会有记忆、语言、认知功能的丧失及人格改变等精神行为症状,最终死于肺部感染、褥疮等并发症。目前,AD的发病机制尚不明确,存在tau蛋白过磷酸化、神经细胞膜代谢功能异常等多种假说。主流理论β-淀粉样蛋白(amyloid βprotein,Aβ)级联学说认为,Aβ在脑内过度聚集沉积产生的老年斑(senile plaque,SP)及其所诱导的一系列病理反应是引发AD的重要环节[1]。医学界基于此学说开展了大量针对Aβ和SP清除的药物研究工作,然而研究中出现的药物无效或严重的不良反应迫使研究人员不得不终止实验[2~4],Aβ级联学说也因此受到质疑。这不仅揭示了AD复杂的发病机制,也表明了深入探索Aβ引发AD的一系列机制的必要性。本文就近年来Aβ参与推进AD病情发展的机制研究进行回顾与探究,以望为进一步集成化、整体性研究AD的Aβ级联机制提供支持。
1 Aβ简介
1.1 Aβ的生成
众多研究证实,脑内Aβ的过度生成是诱发AD的核心因素[5]。因此,维持体内Aβ生成与清除的动态平衡是防治AD的关键。研究表明,Aβ生成与淀粉样前体蛋白(amyloid precursor protein,APP)密切相关[6]。APP属单次跨膜蛋白,主要分布在脑内,其余表达于外周组织器官如肝、肾、皮肤等。生理条件下,APP经非Aβ源性途径(即α-分泌酶途径)代谢:首先,APP由α-分泌酶水解为可溶性的片段sAPPα和羧基端的C83;随后,C83又经γ-分泌酶水解为P3和APP胞内结构域(APP intracellular domain,AICD)(图 1)。因 sAPPα具有神经保护作用,且P3为易被降解的不完整Aβ分子,故该途径对机体不会产生有害影响[7~8],然而,最新研究表明AICD可能介导AD病理学改变[9~10]。病理条件下,与Aβ生成息息相关的肿胀神经轴突内的β-位点剪切酶-1(β-site APP cleavage enzyme-1,BACE-1)活性异常上调[11],这时APP主要经淀粉样变途径(即β-分泌酶途径)代谢:首先,APP经BACE-1酶切割为β-APP和C99;随后,C99又经γ-分泌酶作用生成Aβ40(约90%)和Aβ42(约 10%)两种主要形式(图 1)。其中,Aβ40常易进入脑血管,诱发脑淀粉样血管病,加重AD病理[12],而Aβ42具有很强的疏水性,容易聚集成寡聚体,对轴突和突触产生毒性作用,导致神经元变性坏死及突触功能障碍,诱发AD病理变化[13]。
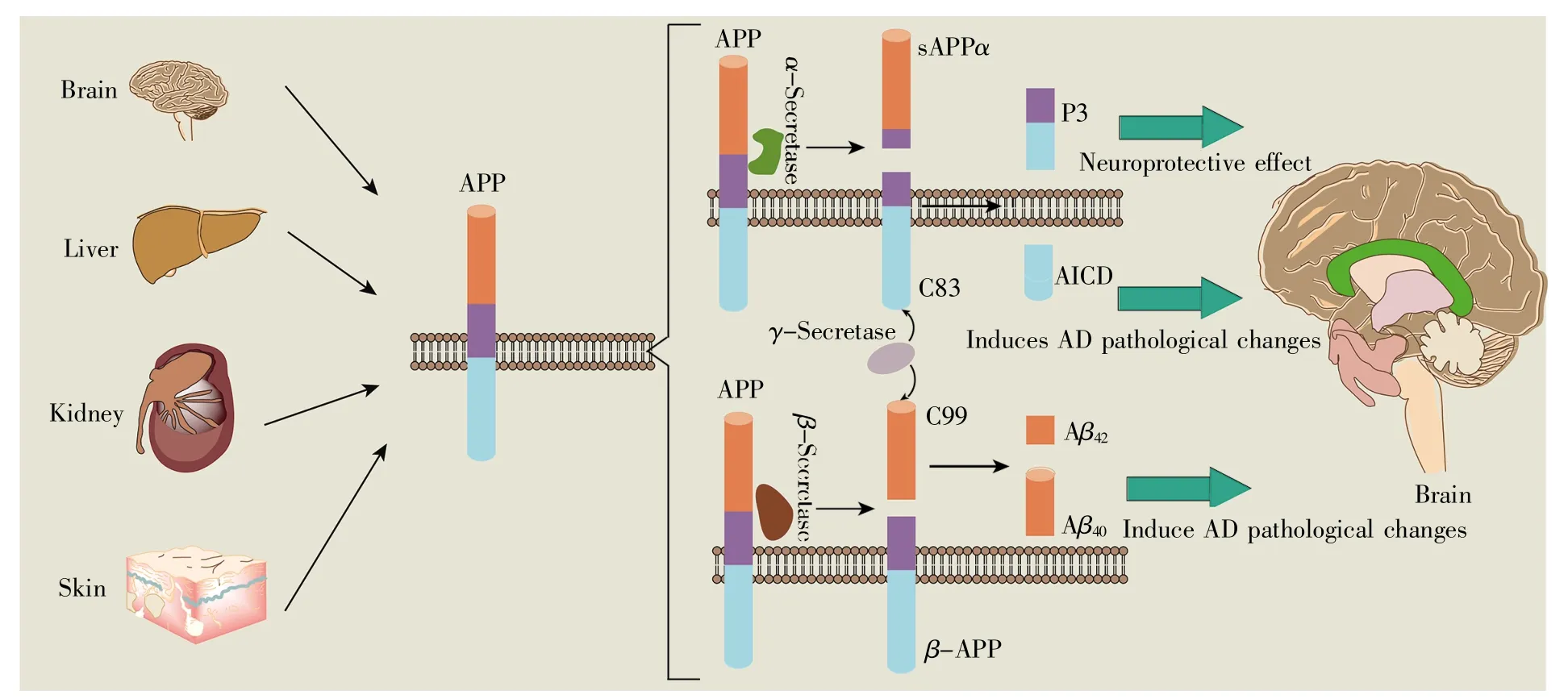
图1 Aβ的生成过程生理条件下,脑、肝、肾、皮肤产生的APP,经α-分泌酶水解为sAPPα和C83,C83又经γ-分泌酶作用产生神经保护性质的P3和诱导AD病理改变的AICD;病理条件下,APP主要经β-分泌酶切割生成β-APP和C99,后者再经γ-分泌酶水解为引发AD病理改变的Aβ40和Aβ42两种主要形式。Fig.1 The production process of AβUnder physiological conditions,APP produced by the brain,liver,kidney,and skin is hydrolyzed by α-secretase into sAPPα and C83,and C83 is activated by γ-secretase to produce neuroprotective P3 and AICD that induces pathological changes in AD.Under pathological conditions,APP is cleaved by β-secretase to generate β-APP and C99,and the latter is hydrolyzed by γ-secretase into Aβ40and Aβ42that cause AD pathological changes.
基因突变是影响APP代谢、生成更多Aβ的重要因素。研究证实,APP基因位点(如670/671位点、711位点、716位点)的改变常会诱发更多BACE-1酶剪切APP,产生高于健康人4~10倍的完整的、游离的Aβ[14]。有研究报道,早老素蛋白-1的突变可能在AD早期阻止APP正常分解或转运,甚至直接改变γ-分泌酶构像,加快γ-分泌酶与C99肽的相互作用,促进长链Aβ生成,增加Aβ42/Aβ40比率[15~16]。此外,载脂蛋白 E(apolipoprotein E,ApoE)突变是散发型AD的高风险因素。相关研究表明,单个ApoEε4基因携带者的AD患病风险增加3~4倍,而一对ApoEε4基因携带者的患病风险将提高至 9~15 倍[17~18]。究其原因,可能与 ApoEε4 和受体结合后,激活非经典丝裂原激活的蛋白激酶(mitogen-activated protein kinase,MAPK)信号转导通路,进而调控APP转录,影响其裂解位点,增加Aβ 水平有关[19]。
1.2 Aβ的清除
生理情况下,机体产生的Aβ可经以下途径清除,以维持体内Aβ生理水平的稳定。1)受体介导的跨血脑屏障转运。晚期糖基化终末产物受体和低密度脂蛋白受体相关蛋白质分别介导Aβ由血入脑、由脑入血的转输,参与维持大脑与外周组织器官Aβ水平的稳定[20~22];2)中枢神经系统的淋巴循环。胶质淋巴系统和脑膜淋巴管是中枢神经系统淋巴循环的重要组成部分,其在清除Aβ等大分子废物上发挥着重要作用[23]。有研究显示,淋巴循环功能受损将导致淋巴交换功能障碍,使得Aβ在脑内大量沉积[24];3)胶质细胞的清除作用。正常情况下,胶质细胞可发挥清除胞内Aβ的作用,但是,Aβ的不断增多则会诱导胶质细胞释放大量炎症因子,引发炎症反应,并介导Aβ生成[25];4)Aβ的降解酶系统。脑啡肽酶(neutral endopeptidase,NEP)、胰岛素降解酶(insulin degrading enzyme,IDE)和内皮素转换酶(endothelin converting enzyme,ECE)等均可清除Aβ,但在AD患者脑内,上述酶的活性常受到抑制,使得Aβ降解减少;5)外周清除途径。Aβ可转运到外周组织和器官经肝、肾、胃肠道和皮肤等代谢,进而降低脑内Aβ水平[26]。因此,上述任一环节的损伤或功能缺失都会造成Aβ在脑内过度沉积,引发AD病理变化[27]。
2 Aβ级联学说与AD的病理关系
当基因发生突变或Aβ清除途径受损时,Aβ生成与清除的动态平衡被打破,大量Aβ聚集在脑内,引发一系列病理反应。例如:Aβ作用于神经元,经多种细胞信号转导途径,诱导细胞大量凋亡[4]。同时,Aβ在突触聚集,损伤突触可塑性,阻断长时程增强效应(long-term potentiation,LTP),降低AD患者学习记忆能力[28]。此外,Aβ的过度堆积可激活胶质细胞,活化后的胶质细胞一方面能直接吞噬突触,造成突触损伤和丢失,另一方面又释放大量炎症介质和氧化因子等有害物质,引发炎症反应及氧化应激,并上调BACE-1活性,诱导过多Aβ生成,形成一个恶性循环,推动AD病情发展[21]。除此之外,过多的Aβ还能加速tau蛋白过磷酸化,诱发神经原纤维缠结(neurofibrillary tangle,NFT),使轴突变性坏死,导致脑组织出现萎缩乃至结构功能完全丧失,最终引发AD[29]。以上为Aβ级联学说的主要内容(图2)。
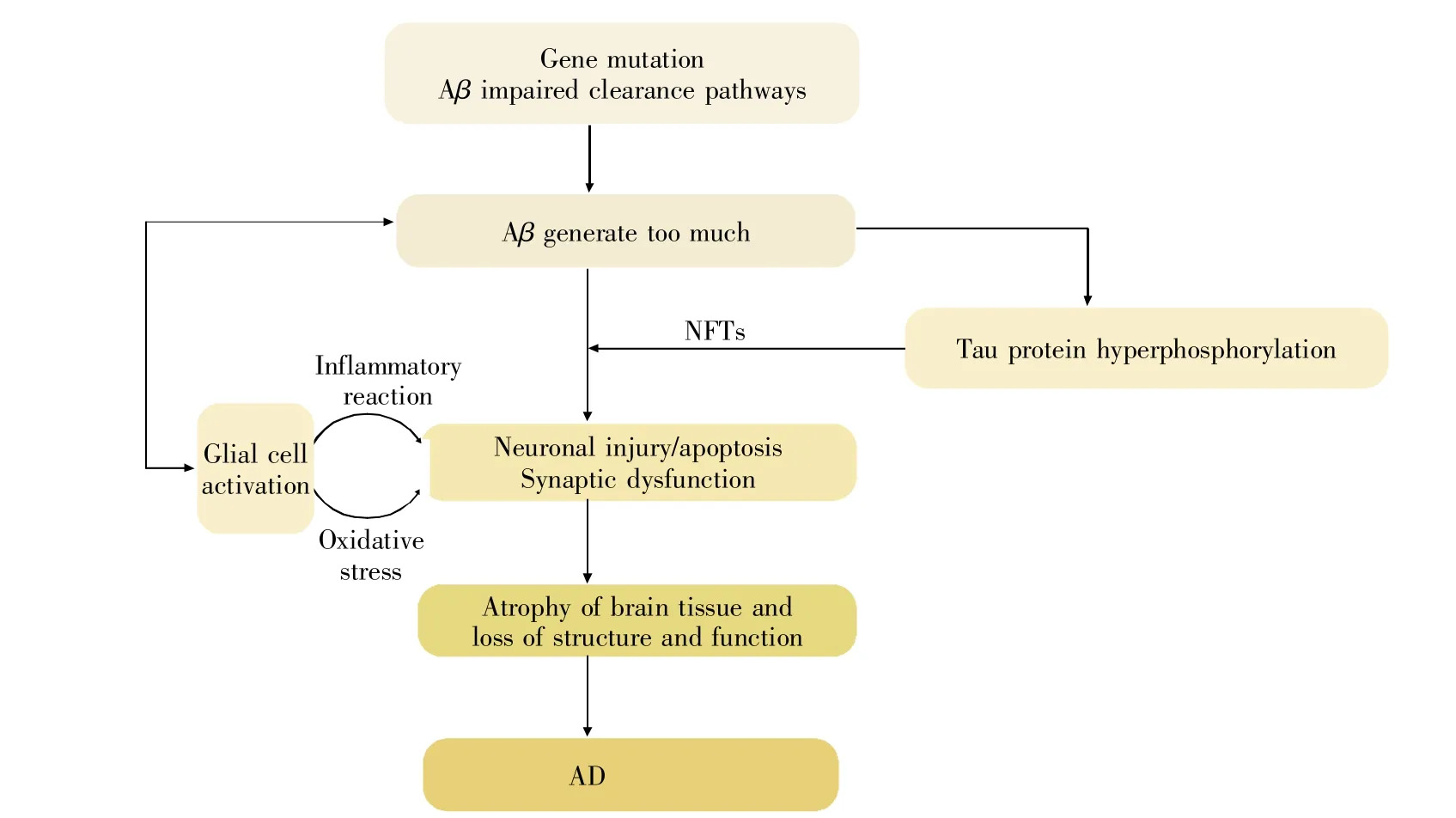
图2 Aβ级联学说主要内容基因突变或Aβ清除途径受损导致Aβ过度生成,大量的Aβ介导神经元损伤、凋亡和突触功能障碍,同时刺激胶质细胞活化。活化的胶质细胞产生炎症反应和氧化应激,从而诱导Aβ大量生成,形成一个恶性循环,加剧神经元损伤和突触功能障碍。此外,Aβ还加速tau蛋白过磷酸化,促进NFTs形成,引发轴突变性坏死,导致脑组织萎缩、结构功能丧失,最终引发AD。Fig.2 The main content of Aβ cascade theoryGene mutation or Aβ clearance pathway is impaired,and excessive Aβ is produced,which mediates neuronal damage,apoptosis and synaptic dysfunction.At the same time,it stimulates glial cell activation,produces inflammatory response and oxidative stress,and induces the massive production of Aβ.A vicious circle aggravates neuronal damage and synaptic dysfunction.In addition,Aβ also accelerates the formation of NFTs by tau protein hyperphosphorylation,triggering axonal necrosis,leading to brain tissue atrophy,loss of structure and function,and ultimately leading to AD.
2.1 Aβ与神经元凋亡
神经退行性病变是诱发AD患者记忆认知功能衰退的重要原因,而神经元的凋亡在其中扮演重要角色。研究发现,Aβ在脑内的积累是诱发神经元凋亡的中心环节,Aβ可参与介导线粒体凋亡通路、死亡受体通路和内质网凋亡通路,三者共同诱导神经元细胞凋亡。
线粒体是细胞动力工厂,为细胞提供必要的能量,参与细胞分化、凋亡等过程,是线粒体凋亡通路的控制中心,而Aβ介导的细胞内钙超载是启动线粒体凋亡通路的关键因子。研究证明,APP裂解产生的一部分Aβ可停留于神经元脂质双层中,并进化成Aβ寡聚物,诱导神经元膜上形成通道样的活性孔,促使钙离子内流增加[30]。同时,Aβ可通过调节钙通道上N-甲基-D-天冬氨酸受体(N-methyl-D-aspartic acid receptor,NMDAR)介导的钙/钙调蛋白依赖性蛋白激酶Ⅱ、钙调磷酸酶、蛋白磷酸酶1及环磷酸腺苷应答元件结合蛋白(cAMP response element binding protein,CREB)等信号通路,诱导细胞内钙超载[31]。钙稳态的失衡,一方面可升高线粒体膜上的促凋亡蛋白Bax(Bcl2-associated X)水平,降低抗凋亡蛋白Bcl-2(B cell lymphoma 2)的表达,使线粒体外膜透化,释放细胞色素c,后者与细胞凋亡蛋白酶激活因子-1、胱天蛋白酶-9(caspase-9)结合成凋亡体复合物,并激活caspase-3,启动caspase蛋白酶级联反应,介导细胞凋亡[32];另一方面可加快Aβ与Aβ结合乙醇脱氢酶的相互作用,诱导活性氧(reactive oxygen species,ROS)等有害物质产生,引发线粒体损伤,并刺激BACE-1酶、γ-分泌酶裂解APP生成更多Aβ,形成恶性循环[33~34]。此外,线粒体融合与分裂的动态平衡是维持其形态、结构功能稳定的重要方式。Aβ刺激会造成线粒体融合蛋白1(mitofusin 1,Mfn1)和Mfn2显著下降,而分裂相关因子高度上调,引起线粒体外膜通透性增强,使细胞色素c大量释放,激活caspase-3,最终触发细胞凋亡程序[35]。
死亡受体是一种胞外富含半胱氨酸区域,且胞质区含有一同源氨基酸构成的死亡结构域(death domain,DD)的特殊蛋白质受体,为细胞外部信号触发凋亡的主要途径。其能够与凋亡信号密切结合,激活凋亡启动子caspase-8,进而活化凋亡执行子caspase-3,触发细胞凋亡反应。Fas/FasL系统是重要的细胞死亡受体凋亡通路。生理条件下,细胞型Fas相关死亡域样白介素-1β转换酶抑制蛋白能够与衔接蛋白分子(Fas-associated death domain,FADD)和caspase-8结合,从而阻止Fas/FasL触发的细胞凋亡程序[36]。然而,AD患者脑内Aβ的大量积累会激活c-Jun氨基端激酶(c-Jun N-terminal kinase,JNK),并增强活化转录因子2的表达,从而上调Fas/FasL水平,促使Fas与其配体FasL大量结合成三聚体,介导胞内DD和FADD聚合,并与caspase-8相互作用生成死亡信号复合物,最终激活caspase-8和caspase-3,启动细胞凋亡程序[37]。此外,有研究显示,Aβ可通过诱导Toll样受体4/6与CD36结合形成复合物,以及核苷酸结合寡聚化结构域样受体蛋白3(nucleotide-binding oligomerization domain-like receptor protein 3,NLRP3)生成,引发炎性反应,介导炎性体衔接蛋白与caspase-8相互作用,从而损伤线粒体[38]。同时,Aβ经ROS/JNK/p53途径上调凋亡因子含量,激活caspase-8,并将Bid裂解为tBid,促进Bax向线粒体转移,引起细胞色素c的释放,从而激活线粒体凋亡通路[39]。因此,死亡受体通路和线粒体凋亡通路是相互联系、不可分割的,共同介导细胞凋亡反应。
内质网(endoplasmic reticulum,ER)参与蛋白质的合成、修饰、加工及转输,胞内未折叠或错误折叠蛋白质的出现,可导致ER功能稳态失衡,形成内质网应激(endoplasmic reticulum stress,ERS),并启动未折叠蛋白质反应(unfolded protein response,UPR),从而停滞大多数蛋白质翻译,增强折叠能力,加快清除错误蛋白质,维持ER内环境的稳态[40]。然而,Aβ的错误折叠过多常会造成持续不可逆的ERS,使ER稳态失衡,触发ER凋亡通路。研究证明,Aβ引发的钙超载,不仅介导线粒体凋亡通路,还会刺激ERS中的经典标志性蛋白质分子糖调节蛋白78与UPR的3个蛋白质受体:蛋白激酶样内质网激酶(protein kinase RNA-like ER kinase,PERK)、肌醇必需酶 1α (inositolrequiring enzyme 1α,IRE1α)和活化转录因子 6(activating transcription factor 6,ATF-6)分离,并相应激活PERK-真核细胞起始因子2α(eukaryotic initiation factor 2α,elF2α)通路、IRE1α 通路及 ATF-6通路,使一系列促凋亡因子(如C/EBP同源蛋白、死亡受体 DR5、JNK、p38 和 IRE1α)上调,并下调抗凋亡蛋白Bcl-2活性,加速细胞凋亡[41]。此外,持续不可逆的ERS还可直接活化ER特异性凋亡因子caspase-12,进而活化下游caspase-9和caspase-3,引发细胞凋亡反应[42~43]。
2.2 Aβ与突触功能障碍
突触是神经元连接靶细胞并向其传递信号的关键部位,主要由突触前膜、突触后膜和突触间隙构成。神经冲动传导时,突触前膜中富含神经递质的突触小泡移动到前膜并释放其中的神经递质,后者通过突触间隙与突触后膜上的受体结合,完成一次信号转导。突触功能受损时,神经冲动传导受阻,从而诱发AD患者记忆认知功能缺陷。Aβ引发的突触兴奋性与抑制性信号转导失衡是突触受损的标志,在Aβ干预神经母细胞瘤细胞的实验中研究人员发现,Aβ可能通过抑制γ氨基丁酸转运体和升高谷氨酸(glutamic acid,Glu)转运体,诱导突触抑制性与兴奋性神经递质紊乱[44]。Glu浓度的升高会加快Aβ与突触可塑性密切相关的NMDAR受体、代谢型谷氨酸受体5(metabotropic glutamate receptor 5,mGluR5)的相互作用,导致NMDAR受体和mGluR5受体激活。NMDAR受体的激活将引发钙离子失调,触发p38 MAPK级联反应,抑制CREB、细胞外信号调控的蛋白激酶(extracellular signal-regulated kinase,ERK)等通路,介导突触可塑性和LTP损伤[45~46];同时又加剧突触线粒体损伤,使ATP合成减少,造成突触功能紊乱。mGluR5受体的激活将介导p38 MAPK、JNK和细胞周期蛋白依赖性激酶反应,抑制LTP,增强长时程抑制(long-term depression,LTD)效应,并改变突触形态,进而加重AD患者记忆认知功能障碍[47]。此外,Aβ还可通过减少脑源性神经营养因子(brain-derived neurotrophic factor,BDNF)的表达,改变突触后树突棘形态,增加细长型而减少短粗型,从而延长树突棘长度,降低其密度,引发突触功能障碍[48~49]。
2.3 Aβ与炎症反应
炎症反应贯穿AD整个病理过程,而Aβ介导的神经胶质细胞活化是引发炎症反应的关键因素。神经胶质细胞是大脑中除神经元外的另一大类细胞,包含小胶质细胞(microglia,MG)、星形胶质细胞(astrocyte,AS)、室管膜细胞和少突胶质细胞,其在维持神经冲动及大脑功能的正常发挥中起着重要作用,但在某些因素的刺激下,则会介导慢性持续性炎症反应,造成神经元损伤和突触功能障碍,引发疾病。
MG是中枢神经系统的免疫防御细胞,能够接触受伤或发育不完整的神经元并吞噬其凋亡碎片,且通过精细的突触剥离移除异常突触,调节突触可塑性,参与维持大脑正常发育及中枢神经系统内环境的稳态。当机体出现病原体或凋亡细胞碎片时,MG被活化为胞体大、分枝稀疏粗大的阿米巴样吞噬细胞形态,并快速迁移到损伤部位,发挥吞噬功能,同时释放肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)、白介素-1β (interleukin-1β,IL-1β)和IL-6等促炎细胞因子,以及趋化因子、ROS和活性氮自由基(reactive nitrogen species,RNS)等物质,募集更多MG和AS加入,参与维持内环境稳态。众多研究表明,MG在AD中具有双刃剑的功能。在AD早期,Aβ的产生可促使MG转化为M1型促炎性和M2型免疫抑制剂两类[50],它们一方面经清道夫受体、CD36、Fc受体、补体C1q受体和补体受体3吞噬Aβ;另一方面产生促炎因子和抗炎转化生长因子-β,保持机体内促炎与抗炎的稳态平衡[51]。随着病情进展,Aβ不断增多,MG过度活化,进而上调基质金属蛋白酶水平,破坏血-脑屏障,使白细胞进入脑内,诱发炎症反应;同时,RNS、ROS等有害物质及TNF-α、IL-1β、IL-6等炎症因子被大量释放,促炎与抗炎稳态平衡被打破,引发慢性持续性炎症反应,导致神经元损伤和大脑功能障碍[52]。此外,Aβ的沉积还会造成线粒体损伤和有丝分裂功能障碍,降低MG吞噬功能并诱发NLRP3/caspase-1依赖性神经炎症,且抑制MG胆碱能抗炎途径和细胞稳态因子髓样细胞触发受体2(triggering receptor expressed on myeloid cells 2,TREM2)及 Toll样因子的表达,加剧炎症反应[50]。
AS是神经胶质细胞中数量、功能最多的一类细胞,能够提供支持框架,固定神经元,并产生促进神经元生长及突触形成的生长因子和乳酸等营养物质,修复神经系统损伤。同时,AS还能通过指引突触或树突的形成位置控制神经元形成,并介导Glu、D-丝氨酸和ATP等神经递质转运,参与维持神经元的信息传递功能。生理状态下,AS可依赖于ApoE脂化,增强MG清除Aβ的能力,或上调NEP、IDE、ECE和激肽释放酶相关肽酶7的表达,从而加快Aβ清除[53~54]。但在AD患者脑内,Aβ的沉积超出了AS的清除阈值,导致AS清除功能障碍并被激活,使其形态学发生改变并大量增殖分化,产生炎症反应。AS通过下调MG中TREM2水平,降低TREM2稳定性,活化MG,并将AS极化为具有神经毒性的表型[55],同时释放IL-6、IL-1β、TNF-α 和干扰素(interferon,IFN)等炎症因子,参与炎症反应;而 IL-1β、TNF-α 和 IFN-γ又可刺激AS上调BACE-1活性,加速Aβ生成,介导慢性持续炎症反应[56]。
2.4 Aβ与tau蛋白过磷酸化
Tau蛋白表达于轴突之中,具有促进微管组装、保持微管结构稳定性的作用,并在维持轴突生长、传递信号冲动及神经元可塑性中都起着广泛作用[57],而过度磷酸化会导致其从微管中解离出来并形成NFTs,诱发神经退行性病变[58]。越来越多的证据表明,Aβ与tau蛋白之间存在密切联系,二者在推动AD演变过程中发挥着特殊作用。在缺乏Aβ的情况下,海马tau蛋白的沉积难以诱发AD神经退行性病变[59],而与之相反的是,在AD早期,tau病理尚未出现时,Aβ就可参与介导神经元损伤及突触形态功能改变,并随着Aβ的不断沉积,其进一步诱导P-tau217和P-tau181释放,加速tau蛋白过磷酸化,引发轴突变性坏死,最终使患者表现为痴呆症状[60]。在Aβ诱导tau蛋白过磷酸化的进程中,糖原合成酶激酶-3β(glycogen synthase kinase-3β,GSK-3β)扮演着重要角色。有研究报道,Aβ激活的凋亡执行子caspase-3除能诱导细胞凋亡外,还能特异性地裂解蛋白激酶B,活化GSK-3β,进而加速tau蛋白过磷酸化;同时,活化后的GSK-3β又能介导BACE-1酶和γ-分泌酶剪切APP,促进更多Aβ生成,形成恶性循环,进一步推动tau蛋白磷酸化[61~62]。此外,细胞型朊蛋白(cellular prion protein,PrPc)与Aβ具有高度亲和力,能够与低浓度的Aβ结合形成Aβ-PrPc复合体并激活酪氨酸蛋白激酶,加速tau蛋白过磷酸化[63]。因此,Aβ与tau蛋白之间并不是简单粗暴的相加模式,而是一种相互协同的神经毒性作用,共同推动AD病程演变[64]。
3 小结与展望
综上所述,基因突变或Aβ清除途径受损造成的Aβ生成增多,不仅通过线粒体凋亡通路、死亡受体通路和内质网凋亡通路触发神经细胞凋亡,还通过提高Glu水平及减少BDNF表达损害突触可塑性,并激活胶质细胞产生炎症反应,同时诱导tau蛋白过磷酸化形成NFTs等一系列病理反应,加剧神经退行性病变和突触功能障碍,以致大脑功能完全丧失和AD发生。近年的研究表明,Aβ在AD中可饰演不同的角色,一方面其作为先天性免疫蛋白,发挥防止真菌、细菌及病毒感染的抗菌肽作用;另一方面感染或无菌性炎症又可驱动 Aβ 沉积,加速 AD 进程[65~66]。因此,Aβ作为诱发AD一系列病理反应的中心环节,减少其表达成为防治AD的有效手段和方法。有研究报道,由渤健公司研发的单抗药物aducanumab能有效进入脑内与Aβ结合,并以剂量依赖性方式降低脑内Aβ沉积,该药物在2021年6月7日被FDA批准用于治疗AD患者[67]。这是基于Aβ级联学说研发的AD新药的成功证明,其不仅给广大患者带去希望,也极大证明了Aβ级联机制在AD发病机制中的合理性[68]。然而,关于AD的Aβ级联机制研究尚存在一定局限性,如:功能障碍的线粒体与内质网凋亡通路之间存在何种联系,线粒体与神经胶质细胞转化之间如何相互影响,等等。诸多问题的解决都需要分子生物医学技术的进步及多学科交融的理论创新来进一步阐明其发病机制。在今后研究中,既要深入探寻各自的调控靶点,更要探索构建Aβ、神经胶质细胞和tau蛋白三者甚至多者之间的相互信号网络,为精准、全面地阐释AD的Aβ级联机制夯实基础,同时也为多靶点药物的设计、开发提供支持[69]。
