TCGA数据库4种消化系统癌症分子特征的比较①
2022-11-16陈世龙王永翠
刘 菲 袁 锐 陈世龙 王永翠
(中国科学院西北高原生物研究所青海省作物分子育种重点实验室,西宁 810008)
消化系统癌症(digestive cancers,DSCs)包括咽喉癌、食管癌(esophageal carcinoma,ESCA)、胃癌(stomach adenocarcinoma,STAD)等消化道癌症和胰腺癌、胆囊癌、肝癌等消化道附属器官癌症,是存在于世界范围内的健康问题[1-2]。其中,ESCA在癌症病死率中排名第六,直结肠癌被认为是DSCs中发病率最高的一种癌症,每年全球新增病例约100万例,死亡55万余人[3-5]。在过去的20年中,结肠癌(colon adenocarcinoma,COAD)、直肠癌(rectum adenocarcinoma,READ)、STAD均为全球发病率最高的10种癌症类型之一。2018年,美国记录了约319 160例DSCs新病例和160 820例与DSCs相关的病死病例,并且DSCs患者的五年生存率均低于20%[6-7]。随着医疗水平的不断提高,美国癌症协会(ACS)、疾病控制与预防中心(CDC)、美国国家癌症研究所(NCI)和北美中央癌症登记处协会(NAACCR)在协作提供的美国癌症发生和发展趋势的年度报告中提出,2011年至2015年间,DSCs的病死率均呈下降趋势,但其整体生存率依然很低[8]。
癌症治疗最终目的是根除癌细胞,同时尽量减少治疗过程中对正常组织的潜在损伤[9]。传统的肿瘤治疗方法是依靠外界力量杀死肿瘤细胞,通常采用的手术治疗方案会对正常组织产生损伤,并且预后效果不好,容易复发,即便患者在接受手术切除前后进行放射治疗或者化学药物治疗,预后仍然很差[10-12]。由于常规治疗方案对转移或复发的DSCs治疗效果很差,寻找其他新型治疗方法必然成为癌症治疗的新趋势[13-16]。近年来,靶向治疗和免疫治疗等肿瘤生物治疗方法已经是相对比较前沿的癌症治疗手段[16]。依据理论,人体正常细胞和肿瘤细胞的分子特征存在很大不同,这些差异可以作为靶点,从而针对已经明确的分子靶点进行靶向治疗[17]。已有大量研究通过机器学习或差异分析的方法揭示一些基因的异常表达或甲基化会影响癌症的发展,并得到一些潜在的癌症预后基因及其通路[18-19]。比如DAVID等[20]同时采用差异分析和机器学习的方法鉴定出多种与肿瘤发展相关的分泌途径有关基因,SAGHALEYNI等[21]则将随机森林的方法用于癌症基因组图谱(cancer genome atlas,TCGA)、基因型组织表达数据(genotype-tissue expression,GTEx)、法国肉瘤组织这3种基因表达数据库中,用以识别新型诊断标志物,并通过实验对其结果进行验证。
尽管之前已有大量的工作对癌症相关的分子数据进行了广泛的研究,但大多数研究都集中于单一分子特征或单个癌症类型之间的关联上。因此,一些学者开始基于癌症集群进行综合分析(泛癌分析),以期发现更系统、更可靠的生物标志物,从而提高预后能力。如HOADLEY[22]等将12种癌症类型进行多平台综合分析,基于基因突变、DNA拷贝数、DNA甲基化、mRNA多组学数据,根据分子特征综合比较12种癌症类型的异同,从而为根据分子亚型对癌症进行分类奠定了基础。此外,BASS等[23]基于体细胞拷贝数分析、外显子分析、DNA甲基化分析、mRNA测序、miRNA测序等方法对295个原发性胃腺癌进行综合分子评估并创建决策树,定义了STAD的4种主要基因组亚型,表现出明显突出的基因组特征,为不同STAD患者相应靶向药物的开发提供指导。不论是在全国还是在全球发布的癌症死亡顺位分布中,人类高发病率和高病死率的恶性肿瘤中均包括READ、肝癌、STAD及ESCA 4种癌症[24]。但目前采用泛癌分析的方法综合分析不同DSCs的多种分子数据的研究依然很少,4种DSCs依然存在一些临床信息及分子特征差异有待于进一步挖掘[25]。
TCGA数据库为癌症研究提供了丰富的资源[26]。利用TCGA数据库的20 000多种原发癌的分子特征和33种癌症类型的正常样本,结合临床数据挖掘不同DSCs的分子特征并加以比较分析,对DSCs的预后具有重要研究价值。因此,本文通过TCGA分析DSCs(ESCA、STAD、READ、COAD)分子数据和临床信息,从而挖掘DSCs分子特征的异同以及临床基本特征的关系。其中,分子数据包括基因突变数据、基因拷贝数变异数据、基因表达数据、基因甲基化数据,所采用的分析方法主要有差异分析、富集分析、生存分析以及相关性分析。本文关于4种DSCs类型异同点的提出可以为深入理解DSCs的分子特征及相应分子靶向治疗方案的设计提供有效依据。
1 资料与方法
1.1 资料 通过TCGAbiolinks包从TCGA公共数据 库 官 网 接 口 获 得DSCs(ESCA、STAD、READ、COAD)分子和样本临床数据,分子数据包括基因突变数据、拷贝数变异数据、基因表达数据以及甲基化数据,样本分为癌症样本和正常样本,正常样本即癌旁组织样本。此外,在TCGA数据库中4种癌症类型患者的临床资料,包括患者的年龄、性别、生存时间、生存状态、癌症阶段分期、发病位置、存活时间。删除生存状态缺失和生存时间不明确的患者数据,临床信息数据最终包括ESCA患者信息185例、STAD患 者434例、READ患者169例、COAD患者458例。
1.2 方法
1.2.1 单一数据类型分析 对每种癌症类型的不同分子数据分别进行数据处理。将基因突变数据采用MaxMIF算法进行筛选,挑选每种癌症类型中30个最大的突变影响函数值(MIF)所对应的基因作为癌症驱动基因[27]。拷贝数变异数据则通过文献报道的显著基因和由COSMIC数据库公布的致癌基因进行筛选,从而分析拷贝数变异情况[16,23,28]。
甲基化数据与基因表达数据做相似处理,对采用log2标准化处理后的基因表达数据通过R Limma包做数据差异分析,为得到包含显著的癌症驱动基因在内的差异表达基因(differential expression gene,DEGs),对数据差异分析采用不同的标准[ESCA:|logFC|>1.2,矫正P值(FDR)<0.05;STAD:|logFC|>1.2,FDR<0.001;READ:|logFC|>1.5,FDR<0.001;COAD:|logFC|>1.5,FDR<0.001],而后将差异分析筛选出的DEGs通过DAVID数据库(https://david.ncifcrf.gov)进行GO和KEGG通路富集分析,-log10P值>2差异具有统计学意义。其中GO分析包括生物学过程(biological process,BP)、细胞组成(cell composition,CC)和分子功能(molecular function,MF)3种注释。对完成在编码区内筛选的甲基化位点数据通过R Limma包做数据差异分析,筛选|logFC|>0.5,FDR<0.001的位点基因为差异甲基化基因(differential methylation gene,DMGs),为得到其GO和KEGG通路,通过DAVID数据库对DMGs进行GO和KEGG通路富集分析,GO分析同样包括生物学过程、细胞组成和分子功能3种注释。
1.2.2 不同数据类型联合分析 将癌症驱动基因的突变数据和基因表达数据分别结合临床信息做生存分析,分析预后基因的预后价值。根据基因在癌症患者中是否发生突变,将癌症患者划分为突变组和未突变组,采用Kaplan-Meier方法,分析癌症驱动基因的突变与4种癌症类型发病之间的关系,从而找到预后效果良好的癌症驱动基因,在对4种癌症类型的癌症驱动基因表达数据结合临床信息生存分析中,将划分组别的标准改为根据基因在癌症患者中表达水平的上下四分位数划分,癌症患者被划分为高表达组和低表达组,分析癌症驱动基因的表达与4种癌症类型发病之间的关系,并采用Survival软件包对结果进行图形可视化,找到具有预后效果的癌症驱动基因。
将突变数据分别与基因表达数据、甲基化数据做联合分析,得到突变与基因表达、DNA甲基化之间的关系。引用皮尔逊相关系数法,利用R软件“cor”函数计算不同癌症类型中癌症驱动基因的突变率,分别与癌症驱动基因的最大表达量、癌症驱动基因甲基化数据最大β值的相关性,并采用ggplot2软件包对结果进行图形可视化。
1.2.3 验证 从TCGA官方发布的文献中找到通过Mutsig计算得到的显著基因[16,23,28],将已报道的显著基因与临床数据结合做生存分析,根据基因在患者中是否发生突变,将癌症患者划分为突变组和未突变组,采用KM方法,分析显著基因的突变与4种癌症类型发病之间的关系,从而找到预后效果良好的显著基因。并将已报道的显著基因突变数据分别与基因表达数据以及甲基化数据利用R软件“cor”函数做相关性分析。将显著基因的一系列分析结果与癌症驱动基因的分析结果做比较。
2 结果
2.1 单一数据类型分析结果 在对ESCA突变数据的筛选中,采用MaxMIF算法计算分别得到4种癌症类型的30个癌症驱动基因,并得到癌症驱动基因在癌症的突变数据中的大体分布(图1)。利用t检验法计算不同癌症类型中每种癌症驱动基因的突变频率,判断每两种癌症类型之间突变频率的差异显著性,经计算得出,两两癌症类型之间P值均<0.001,具有极显著性差异。另外,对各种癌症类型的癌症驱动基因分别做预后分析,得到预后效果显著的癌症驱动基因,其中,ESCA中通过突变数据得到具有预后效果癌症驱动基因为ISG20、IL1R1,SATD癌症驱动基因中具有预后效果的癌症驱动基因为RAB13、SELL,READ癌症驱动基因中预后效果良好的癌症驱动基因为C5,COAD癌症驱动基因中预后效果良好的癌症驱动基因为CD226(图2)。
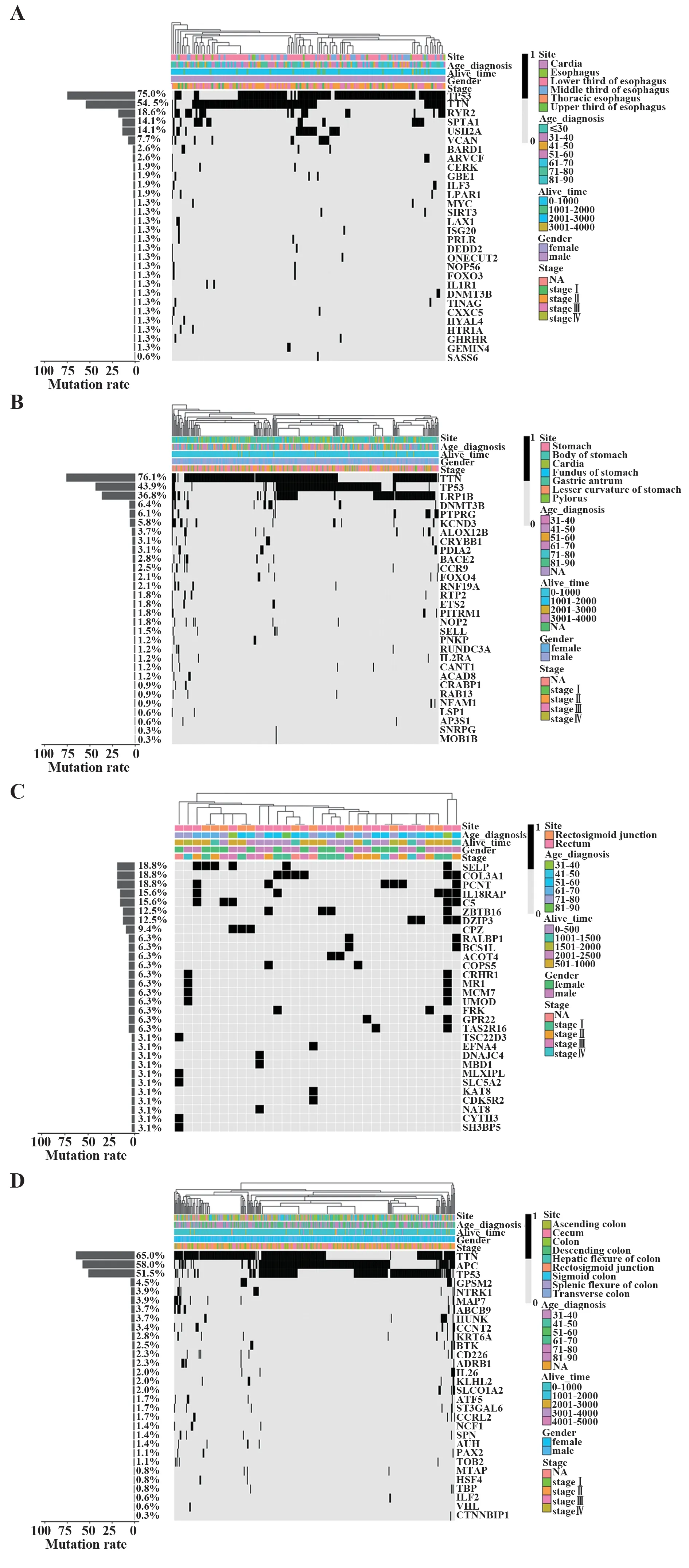
图1 癌症驱动基因突变信息分布及样本信息分布Fig.1 Distribution of sample informations and mutation informations based on cancer driver genes

图2 4种癌症类型的癌症驱动基因预后分析Fig.2 Prognosis analysis of cancer driver genes from four types of cancer
通过癌症驱动基因筛选拷贝数变异数据,观察癌症驱动基因的拷贝数变异情况。将样本分为肿瘤组织样本与正常组织样本,再按基因拷贝数的正负将癌症驱动基因分别分为拷贝数增加和减少2种,以此得到驱动基因拷贝数的4种类型:肿瘤样本中增加、肿瘤样本中减少、正常样本中增加、正常样本中减少。在4种癌症类型中,癌症组织样本和正常组织样本的个数占总样本量的比值均接近于50%,而每种基因在癌症组织样本和正常组织样本中的增加和减少的样本分布并不均匀。针对文献报道的显著基因以及COSMIC数据库公布的常见致癌基因(BRAF、CDKN2A、CTNNB1、EGFR、ERBB2、KIT、KRAS、MYC、PIK3CA、RB1、RET、SMAD4、TP53)统计拷贝数变异的情况(图3)[16,23,28]。可以看出,在不同癌症类型中关键基因的拷贝数变化在癌症组织样本和正常组织样本中的分布并不均匀。
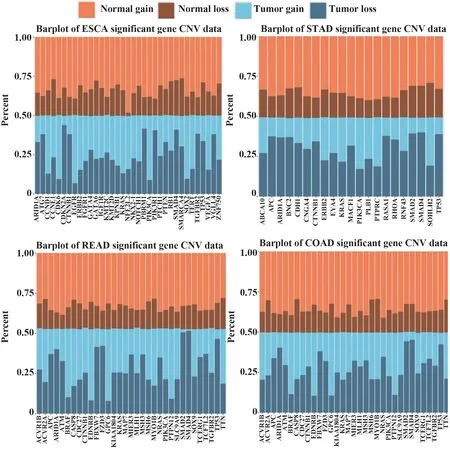
图3 癌症驱动基因的拷贝数变异数据分布情况Fig.3 Distribution of copy number variation data of cancer driver genes
癌症基因表达数据和癌症甲基化基因数据分别通过差异分析筛选,在癌症基因表达数据中筛选到829个ESCA DEGs、394个STAD DEGs、606个READ DEGs、591个COAD DEGs。在癌症甲基化数据中筛选到88个ESCA DMGs、36个STAD DMGs、515个READ DMGs、127个COAD DMGs。对所得到的不同癌症类型DEGs通过DAVID进行KEGG功能富集分析,进一步了解DEGs的富集通路。KEGG富集通路显示,与基因表达相关的差异基因显著富集在神经活性配体与受体相互作用方面(图4)。另外,通过上下四分位数将表达数据分为高表达组和低表达组,并以此为分组做预后分析,得到ESCA中具有预后效果的基因有29个,STAD中具有预后效果的基因有70个,READ中具有预后效果的基因有71个,COAD中具有预后效果的基因有50个。

图4 DEGs KEGG功能富集分析Fig.4 KEGG functional enrichment analysis of DEGs
2.2 利用不同数据类型联合分析的结果 将突变数据分别与基因表达数据做相关性分析,以不同的癌症类型中癌症驱动基因的突变率分别与癌症驱动基因的最大表达量以及甲基化最大Beta值做相关性分析,其中ESCA基因突变率与基因表达量、甲基化Beta值的相关系数r为0.110、0.091,STAD基因突变率与基因表达量、与甲基化Beta值的相关系数r为-0.062、0.057,READ基因突变率与基因表达量、甲基化Beta值的相关系数r为0.200、0.110,COAD基因突变率与基因表达量、甲基化Beta值的相关系数r为0.065、0.018。相关系数的绝对值均<0.300。
通过突变数据做预后分析从TCGA官方发布的文献中找到的显著基因,并将显著基因的突变率分别与基因表达数据、甲基化数据做相关性分析,以此验证MaxMIF算法找到的癌症驱动基因预后率与其他分子数据相关性分析的准确性。ESCA中显著基因32个,STAD中显著基因23个,READ中显著基因28个,COAD中显著基因个数28个,具有预后效果的显著基因所占的比例分别为9.375%、13.043%、0、7.143%,而具有预后效果的癌症驱动基因所占的比例分别为3.333%、6.667%、3.333%、6.667%。预后基因比率差别不大,但通过癌症驱动基因分析得到的潜在预后基因个数稳定一些。显著基因的突变率与基因表达数据相关系数分别为0.008、0.172、-0.031、-0.130,显著基因的突变率与甲基化数据相关系数分别为-0.232、-0.187、0.081、0.220。同样的,显著基因与其他分子数据的相关系数也均<0.300。
3 讨论
DSCs是常见的恶性肿瘤,由于早期无明显不良状况,诊断率低,发现时多濒临或处于中晚期,诊断费用高、治疗疗效差,并伴有易转移、易复发的危险,因此发病率、病死率高,另外,我国DSCs的发病人群也日趋年轻化[28-31]。在最近的10年,随着高通量测序技术的出现、测序成本的大幅度降低以及基因组学、转录组学、蛋白组学、表观组学等技术的进步,医疗大数据得以迅速发展。大规模的研究已经绘制了基因组调控网络,并极大地增进了对包括癌症发生和癌症转移在内的多种生物学过程的分子机制的理解[32]。
已有证据表明,有些基因在多种癌症组织中起着相似作用。例如,基因TP53通过影响NOX4表达与多种癌症的进展建立关系,包括ESCA、甲状腺癌、膀胱尿道癌等,并可调节胰腺癌细胞对化疗药物、信号转导抑制剂以及营养素的敏感性[33-34]。此外,癌症分析还发现不同癌症之间类似的分子特征变化,例如MCM8基因在STAD、READ、肝癌、肺癌、宫颈癌、卵巢癌、淋巴瘤、头颈部和中枢神经系统肿瘤中过度表达,ESM1基因在12种癌症类型中过度表达并与宫颈鳞癌和腺癌、ESCA、肾透明细胞癌和肾乳头状细胞癌4种癌症患者的总生存期显著相关[35-37]。
本研究通过检索TCGA数据库发现,每种癌症类型都具有特有的潜在预后基因,如ESCA中的ISG20、IL1R1,STAD中的RAB13、SELL,READ中的C5、GPC6、EDNRB,以及COAD中的CD226。ISG20基因是由GONGORA等[38]作为早幼粒细胞白血病核体(PML-NB)相关蛋白首先引入的。ISG20的上调表达可以促进肿瘤转移和血管生成,并与肝细胞癌患者的无疾病复发存活期较短呈显著相关,另外,ISG20可以促进局部肿瘤免疫,可能成为人类神经胶质瘤的潜在治疗靶点[39-40]。由IL1R1编码的具有中继信号作用的IL-1Rα与IL-1结合,后IL-1Rα与IL-1受体辅助蛋白形成复合物,诱导了多种通路的激活,并且这些通路都是肠道肿瘤发生的关键[41-43]。RAB13通过多种机制影响细胞迁移,在集体迁移潜在部位也存在丰富的RAB13 RNA,从而为针对集体癌细胞入侵提供有效的打击机会[44]。SELL基因翻译产生L-选择蛋白,是一种细胞黏附分子,在调节免疫细胞运输中起重要作用,并且已经被发现其表达在几种人类癌症中被上调,如SELL上调表达与炎症性微环境以及乳腺癌的良好预后有关[45]。补体系统是先天免疫系统的重要组成部分,在适应性免疫的激活和调节中起着关键作用,而C5是末端补体途径中补体级联的一个组分,补体激活导致C5裂解成小C5a分子和大C5b分子,C5a分子具有免疫刺激作用,而C5b分子是膜攻击复合物(MAC)的初始成分[46-47]。CD226是在各种免疫细胞上表达的激活受体,充当共刺激物,增强T细胞或自然杀伤细胞的活化,并在先天性和适应性免疫调节网络中显示出重要意义[48-49]。可见不同癌症类型中的预后效果良好的基因大多可能与肿瘤免疫方向有关。
对DEGs进行KEGG富集通路分析,得到在神经活性配体与受体相互作用方面显著富集的结果,表明DEGs可能通过神经活性配体-受体相互作用途径在4种DSCs中发挥作用。先前有研究表明,该途径似乎在同为DSCs的肝癌进展中起关键作用,在肝癌的每个阶段都能观察到神经活性的配体-受体相互作用[50-51]。另一项研究说明,神经活性配体-受体相互作用途径参与了肿瘤微环境和细胞间的通讯,其失调可能促进了癌症的发展[52]。
综上所述,本研究报告了ESCA、STAD、READ、COAD 4种DSCs在多组学层面的综合分析。4种癌症类型具有相似的分子特征,包括:4种癌症类型的突变频率与基因表达、甲基化水平无明显相关性;MaxMIF软件获得的癌症驱动基因中均仅有少数几个基因具有良好的预后特征,如ESCA癌症驱动基因中的ISG20、IL1R1,STAD癌症驱动基因中的RAB13、SELL,READ癌症驱动基因中的C5,以及COAD癌症驱动基因中的CD226;拷贝数变化在癌症组织样本和正常组织样本中的分布并不均匀;DEGs中均仅有少部分基因具有良好的预后特征,且KEGG通路都富集在“神经活性的配体-受体相互作用”这条通路上。同时,获得4种癌症类型不同的分子特征,即4种癌症类型的癌症驱动基因、预后良好的DEGs、DMGs均不同。总之,以上结果的提出有助于了解整个DSCs的异同,并有望为深入理解DSCs的分子特征及相应的分子靶向治疗方案的设计提供有效的依据。
