毛发中甲基苯丙胺、苯丙胺、6-单乙酰吗啡及吗啡的检测阈值评估
2022-11-12施晓露车鑫锋吴健美乔宏伟王优美
施晓露 ,车鑫锋 ,吴健美 ,狄 斌 ,乔宏伟 *,王优美 **
(1中国药科大学药学院,南京 210009;2国家禁毒委员会办公室中国药科大学禁毒关键技术联合实验室,北京 100193;3公安部禁毒情报技术中心,毒品监测管控与禁毒关键技术公安部重点实验室,北京 100193)
与常用检材血液、尿液相比,毛发中的毒品检测具有取样简便、易于存贮、检测窗口期长、生长速度稳定等特点[1-3]。通过毛发检测技术可获取更长时程的摄毒信息,推测药物摄入时间,分析用药行为[4-6]。该技术已被广泛应用于公安机关的禁毒工作中,检测结果在吸毒案件认定中具有重要意义[7-8]。
2018年10月,公安部发布的《涉毒人员毛发样本检测规范》(以下简称《规范》)中,根据毛发检测协会(Society of Hair Testing,SoHT)提供的推荐阈值,规定了甲基苯丙胺(MA)、苯丙胺(AM)、6-单乙酰吗啡(6-MAM)、吗啡(MOR)的检测阈值为0.2 ng/mg[9]。根据《规范》的相关规定,当毛发中相关物质的检测含量在阈值以上时,即表明被检测人员在最近6个月内摄入过毒品。
然而,在实际案件中,存在吸毒人员承认吸毒行为但毛发检测含量低于《规范》规定的阈值的情况。一方面,可能是由于各地实验室的前处理方法提取不充分;另一方面,可能是吸毒人员吸食量较低,或者近年来吸毒人员毛发中毒品含量发生较大变化所致。该情况的发生会导致禁毒部门的执法力度无法得到保证。由此可以合理推断,毛发前处理技术应进行系统优化,毛发检测阈值应适当下调。但毛发检测阈值也不能过低,因为非涉毒人员(如毒品实验室技术人员、滥用环境中未吸食人员等)若长期暴露于毒品环境当中,毒品及其代谢物也会存在于毛发中[10],可能造成假阳性的结果。因此,在阈值评估时需要考虑两者之间的平衡,以免造成误判,增加执法成本,影响公安机关的执法公信力。
目前,国内针对如何区分吸毒人群和被动污染人群的阈值研究未见报道。国外相关研究常采用污染实验来制备受污染样品,但这似乎无法反映被动暴露期间实际发生的情况[11-12]。基于以上背景,本研究首先利用UPLC-MS/MS建立了针对甲基苯丙胺、6-单乙酰吗啡及其代谢物快速高效前处理方法,对真实吸毒人员毛发样品和被动接触毒品人员毛发样品分别进行检测分析。通过接受者操作特性曲线(receiver operating characteristic curve,ROC 曲线)、Youden 指数并结合实际应用涉及的影响因素进行阈值评估,同时还分析了两种毒品与代谢物之间的比率关系,旨在探讨一种更加符合我国国情的研判标准,以区分被动污染与主动吸食毒品的人群,为公安部今后修订毛发检测阈值奠定基础。
1 材 料
1.1 药品与试剂
MA、AM、6-MAM、MOR 对照品(公安部禁毒情报技术中心,国家毒品实验室);氘代内标储备液MA-D5、AM-D5、6-MAM-D3、MOR-D3(公安部第三研究所);甲酸铵(质谱纯,美国Sigma-Aldrich 公司);甲醇、乙腈(色谱纯,德国Merck 公司);甲酸(质谱纯,美国Honeywell 公司);十二烷基磺酸钠(化学纯,国药集团化学试剂有限公司);超纯水由Milli-Q超纯水系统(美国Millipore公司)制备,其他试剂均为市售分析纯。
1.2 仪 器
ACQUITY UPLC®Xevo TQ-XS 超高效液相色谱-质谱联用仪(美国 Waters 公司);CPA224S 万分之一电子天平(德国Sartorius 公司);研磨仪(北京万孚智能科技有限公司);KQ-600DE 超声仪(昆山市超声仪器有限公司);MIX-2500 涡旋仪(杭州佑宁仪器有限公司)。
2 方 法
2.1 UPLC-MS/MS分析条件
2.1.1 色谱条件色谱柱:Waters ACQUITY BEH C18(2.1 mm × 100 mm,1.7µm);流动相:A 相为5mmol/L甲酸铵-0.1%甲酸水溶液,B相为乙腈;梯度洗脱程序:0.0~0.5 min,5%B;0.5~1.0 min,5%~10%B;1.0~5.0 min,10%~15%B;5.1~6.0 min,15%~100%B;6.0~6.1 min,100%~5%B;6.1~7.0 min,5%B。流速:0.4 mL/min;进样量:1µL;柱温:40 ℃;样品室温度:4 ℃。
2.1.2 质谱条件 扫描方式:电喷雾(ESI)离子源,正离子扫描;检测方式:多反应监测(MRM);毛细管电压:1 000 V;锥孔电压:20 V;离子源温度:150 ℃;碰撞气流量:0.15 mL/min;脱溶剂气温度:500 ℃;脱溶剂气流量:1 000 L/h;气帘气流量:150 L/h;各目标物MRM参数见表1。

Table 1 MRM parameters of four target compounds with their internal standards
2.2 对照品溶液配制
一级储备液:将4 种目标物用甲醇分别配制成1 mg/mL 的标准品溶液,各取0.5 mL 于10 mL量瓶中,用甲醇溶液稀释成含各目标物50 µg/mL的一级储备液;分别取4 种氘代内标的1 mg/mL 标准品溶液20 µL 于20 mL 量瓶中,用甲醇稀释至刻度,配制成含4 种氘代内标的1µg/mL 的一级储备液。
工作内标溶液:取氘代内标一级储备液1 mL于50 mL 量瓶中,用甲醇稀释至刻度,得20 ng/mL氘代内标混合溶液。取适量,按甲醇与水体积比7∶3配制成14 ng/mL工作内标溶液。
2.3 样品前处理
取适量毛发于15 mL塑料离心管中,依次加入0.1% SDS 溶液、纯水、丙酮各5 mL 振荡洗涤后置于通风橱中晾干。称取毛发样品约20 mg 于5 mL具盖研磨管中,分别加入甲醇-水(7∶3)溶液和工作内标溶液500µL。在研磨仪中以3 000 r/min 研磨提取100 s。提取完成后,用1 mL 一次性注射器移取液体并过0.2µm GHP 微孔滤膜于2 mL 离心管中。取适量过滤后的提取液与甲醇-水(3∶7)溶液按体积比1∶1混合至进样小瓶中,涡旋仪混匀后使用 UPLC-MS/MS分析[13]。
2.4 方法学验证
2.4.1 选择性 将不同来源的空白毛发、加标空白毛发及同时加入混合对照品与内标的空白毛发,经“2.3”项下前处理后检测。考察空白毛发基质与工作内标溶液是否影响对目标分析物的测定。图1 显示空白毛发基质和工作内标溶液对各目标物的检测无明显干扰,该分析方法可以区分目标分析物与基质中的其他组分。

Figure 1 Chromatograms of four target compoundsA: Blank hair samples containing 20 ng/mL working solution of 4 target compounds;B: Blank hair samples with internal standards of 4 target compounds;C:Blank hair matrix of 4 target compounds
2.4.2 线性范围、检出限及定量下限 取适量4种目标物的一级储备液用甲醇-水(7∶3)溶液配制成系列浓度梯度的混合工作溶液,各取500 µL 加入空白毛发中,再加入工作内标溶液500µL,按照“2.3”项前处理后得到质量浓度为0.01,0.02,0.05,0.1,0.2,0.5,1,2,4,5 ng/mg 的标准曲线对照品毛发样本,进行分析。以目标物的峰面积(y)为纵坐标,与之对应的浓度(x)为横坐标作线性回归(权重:1/x)。同法将一级储备液加入到空白毛发中,配制成最终质量浓度为0.001,0.002,0.004,0.005,0.008 ng/mg 的毛发样本进样分析,以离子对丰度比符合规定范围且信噪比S/N=3为检出限(LOD),S/N=10为定量下限(LLOQ)。AM在0.01~4 ng/mg 的范围内线性良好,其他目标物在0.01 ~5 ng/mg 范围内线性良好。LOD 在0.001 ~ 0.008 ng/mg,LLOQ 均为0.01 ng/mg。决定系数(R2)均大于0.999 6,结果见表2。

Table 2 Linear ranges,determination coefficients,lower limit of detection(LLOQ)and limit of detection(LOD)of four target compounds
2.4.3 准确度和精密度 将4种目标物的一级储备液用甲醇-水(7∶3)溶液逐级稀释为一系列混合工作溶液,按照“2.3”项下各取500µL加入空白毛发中,再加入工作内标溶液500µL经前处理后,配制为低(0.02 ng/mg)、中(0.5 ng/mg)、高(4 ng/mg)3 个浓度的对照品毛发样本,每一浓度点各制备6个平行质控样品,于“2.1”项下分析方法进行分析,连续测定3 d,计算准确度、日内精密度和日间精密度,各分析物准确度和精密度见表3。
2.4.4 提取回收率和基质效应 将4种目标物溶液使用甲醇-水(7∶3)溶液作为稀释剂配制成低(0.4 ng/mL)、中(10 ng/mL)、高(80 ng/mL)3 个浓度,每个浓度平行配置6 份,与等体积工作内标溶液一同加入装有空白毛发的研磨管中,经前处理后进样分析,记录峰面积为(Ab);同法配制上述3 组目标物溶液,加入内标后调节甲醇与水体积比为1∶1 后进样分析,记录峰面积为(Aa);使用毛发基质溶液作为稀释剂配制3 组目标物溶液,加入内标后调节甲醇水比例为1∶1 进样分析,记录检测峰面积记为(Ac)。基质效应=Ac/Aa;提取回收率= Ab/Ac。由表3 可知,基质效应为99.19% ~109%,说明毛发基质对检测无明显干扰。各待测物的提取回收率均在85%~115%以内。
2.5 方法应用
2.5.1 样本信息 2020 年采集社区戒毒社区康复人员毛发281 份和各地公安机关毒品实验室收集的吸毒人员毛发242 份,共计523 份。采集自各地毒品实验室技术人员的毛发共430 份,毒品实验室技术人员的样本作为易被动污染样本,用于代表社会层面有可能因为被动污染而被误判的人群,同时也能较好地模拟实际暴露污染的情况。
2.5.2 检测结果 表4汇总了两组分析对象毛发中4种目标物的检测统计数据。
吸毒人员组:在高于定量限的443 份MA 样本中,有133 份(30.0%)样本未同时检测到AM,这133 份样本中检测到的 MA 含量有101 份(75.9%)低于0.1 ng/mg。在高于定量限的198份6-MAM 样本中,有19份(9.6%)样本未同时检测到MOR。
毒品实验室技术人员组:在检测到高于定量限的 67 份MA 样本中,有 57 份(85.1%)样本未同时检测到AM,这57 份样本中检测到的MA 含量有53份(93.0%)低于0.1 ng/mg。在高于定量限的24份6-MAM 样本中,有20 份(83.3%)样本未同时检测到MOR。

Table 4 Detection of four target compounds in drug intaking group and contamination group
表5 统计了吸毒人员毛发中代谢物与原形药物的比率。吸毒人员组:310 份(70%)同时检测到高于定量限的 MA 和 AM 样本中,AM 与 MA 的比率为0.000 5~0.359 7。179 份(90.4%)同时检测到高于定量限的 6-MAM 和MOR 样本中,MOR 与6-MAM 的比率为0.048 4~7.929 8。毒品实验室技术人员组:10 份(14.9%)同时检测到高于定量限的 MA 和 AM 样本中,AM 与 MA 的比率为 0.008 ~0.159。4 份(16.7%)同时检测到高于定量限的6-MAM 和 MOR 样 本 中 ,MOR 与 6-MAM 的 比 率 为0.13~0.22。
2.6 统计分析
金标准:吸毒人员组中承认吸食毒品的人员相应的毛发检测含量为阳性样本,毒品实验室技术人员作为易接触毒品人群,相应的目标物毛发检测含量为阴性样本。吸毒人员组承认吸食甲基苯丙胺的受试者有379 名、承认吸食海洛因的有216名。
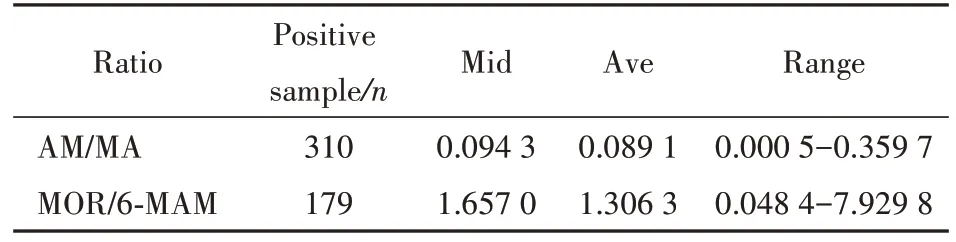
Table 5 Metabolite-to-parent drug ratio of drug users
Kruskal-Wallis H 检验和 Mann-Whitney U 检验结果显示,甲基苯丙胺379 份阳性样本和430 份阴性样本、海洛因216 份阳性样本和430 份阴性样本之间有显著性差异(P < 0.05)。
4 种目标物绘制的ROC 曲线如图3 所示。MA、AM、6-MAM、MOR 的 AUC 分 别 为 0.948,0.927,0.840,0.863。根据评价指标,当ROC 曲线的曲线下面积AUC =[0.85,0.95],说明此分类器性能很好。通过本检测方法的定量限制约及最高Youden 指数所对应的阈值,得到理论最佳判定阈值分别为:MA ≥0.018 ng/mg,特异性和灵敏度为90.5%、91.6%;AM ≥ 0.010 ng/mg,特异性和灵敏度为90.5%、91.6%;6-MAM ≥ 0.036 ng/mg,特异性和灵敏度为97.4%、67.6%;MOR ≥ 0.011 ng/mg,特异性和灵敏度为99.3%、72.1%。该阈值要远低于现行《规范》中规定的阈值。具体统计数据详见表6。

Figure 3 Receiver operating characteristic (ROC) curves of four target compoundsA:MA;B:AM;C:MOR;D:6-MAM
3 讨 论
3.1 前处理方法优化
提取溶剂的优化:对甲醇、乙腈、超纯水、甲醇-水(1∶1)和乙腈-水(1∶1)5 种提取溶剂的提取效果进行考察[14]。结果显示,甲醇-水和乙腈-水较好,但由于乙腈-水会使MOR 在色谱检测中裂峰,而甲醇-水与乙腈-水的提取效果相当,因此最后选择甲醇-水作进一步的优化。分别考察了甲醇-水体积比为1∶9~9∶1的提取效果,体积比为7∶3时即提取效果最优。因此选择甲醇-水(7∶3)作为提取溶剂。
同时,对进样溶剂比例、研磨转速、研磨时间、超声时间也进行了优化。以甲醇-水(7∶3)的溶剂比例直接进样检测会导致MOR 裂峰,故经试验后调节溶剂最终进样比例为5∶5 以优化MOR 峰型;磨仪转速的增加会使研磨更充分,但高转速下会使提取剂升温、膨胀导致研磨管损坏,根据结果综合考虑最终选择3 000 r/min;由于各目标物浓度基本在100 s 时达到峰值,故最终选择研磨时间为100 s;超声处理能够有效地降低毛发的结构强度,提高提取效率,但结果显示超声处理的增效作用并不明显,因此,为了缩短前处理时间选择不进行超声处理。
3.2 阈值评估
阈值评估的目标是使真阳性率(灵敏度)尽可能高,以识别到更多的吸毒人员,同时要使假阳性率尽可能低,以免造成误判。因此引入了Youden指数计算理论最佳阈值。美国滥用物质和精神健康服务管理局(Substance Abuse and Mental Health Services Admission,SAMHSA)规定,实验室应该有在阈值的40%浓度时的定量分析能力[15]。受限于目前检测技术的水平,除6-MAM 的理论最佳阈值0.036 ng/mg 能够满足SAMHSA对实验室的定量要求外,MA、AM、MOR的阈值暂时都无法直接应用。
理论最佳阈值是以假设其假阴性和假阳性的危害性具有同等意义为前提下Youden指数最大时所对应的值,但由于毒品检测的特殊性,在实战应用时两者危害性并不相等。不能单纯以理论最佳阈值作为这4种目标物的判定阈值,除了参考AUC与Youden 指数外,还需要对执法成本和执法力度进行抉择,针对当下情况选择一个更加符合实际的阈值。表7 给出了不同阈值下的真阳性率和假阳性率作为参考。根据表7,MA 在以0.1 ng/mg 作为阈值时,AM 在以0.025 ng/mg 作为阈值时,MOR在以0.05 ng/mg 作为阈值时,真阳性率有较大提升,假阳性率也维持在较低水平,而6-MAM 的阈值在0.025与0.05之间真阳性率变化不大,但假阳性率变化明显,因此将阈值定为0.05 ng/mg,故最终推荐阈值为:MA ≥ 0.1 ng/mg,AM ≥ 0.025 ng/mg,6-MAM ≥ 0.05 ng/mg,MOR ≥ 0.05 ng/mg。但受限于阴性组样本量、仪器检测水平和执法成本与力度之间的取舍问题,本研究给出的建议阈值,在未来研究中仍有较大的可优化空间。

Table 6 Statistical results of ROC curve analysis of four target compounds
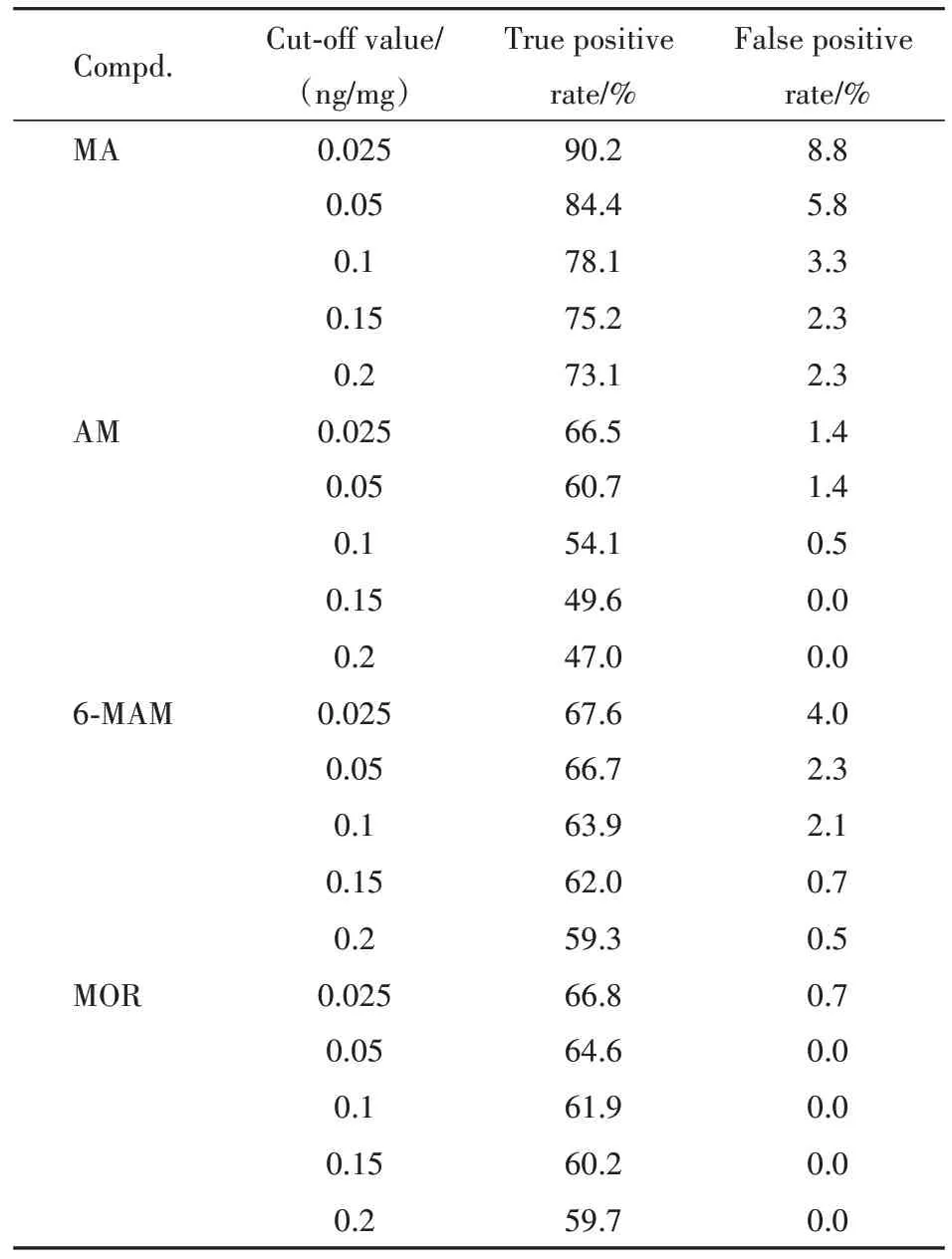
Table 7 Reference ranges for four target compounds cut-off value
代谢物与原形药物比率在判断被动污染与主动吸食时也具有一定意义[16-17]。但由于个体差异、吸毒习惯等不确定性因素的影响,该比率范围较宽且不唯一,可作为区分主动吸食与被动污染的辅助参考指标,不充当必要条件。根据试验结果可以发现,阴性组的比率范围要明显低于阳性组,并且阳性组与文献中报道的比率范围相符[18-19]。
4 总 结
基于公安禁毒部门在执行《规范》过程中发现的实际问题,本研究首先优化了毛发中AM、MA、6-MAM、MOR 的前处理方法,该方法符合生物样本分析的各项要求,且能够稳定、准确、快速,可靠地运用于实际涉毒案件中。
应用本研究优化的前处理方法,对收集自全国各地吸毒人员与毒品实验室技术人员毛发样品进行分析。基于分析结果得出的理论最佳阈值低于《规范》中建议的阈值。经过与实际相结合重新评估,本研究提出的推荐阈值,能为公安系统对阈值的合理评估提供参考。
