高粱千粒重全基因组关联分析和候选基因预测
2022-10-29邹桂花丁延庆徐建霞陈合云刘合芹郑学强张立异
邹桂花 丁延庆 徐建霞 曹 宁 陈合云 刘合芹 郑学强 张立异,*
(1 浙江省农业科学院作物与核技术利用研究所/浙江省数字旱粮重点实验室,浙江 杭州 310021;2 贵州省农业科学院贵州省旱粮研究所,贵州 贵阳 550006)
高粱[Sorghumbicolor(L.)Moench]是我国最早栽培的禾谷类作物之一,因其具有很强的抗逆性和适应性,在全国各地均有种植。历史资料表明,高粱在解决粮食问题中曾发挥过巨大作用[1]。随着水稻、小麦、玉米等粮食作物育种水平的提高以及人民生活条件的改善,高粱的利用价值发生了根本性转变,由曾经的口粮变成了如今的工业原料,用于酿酒、饲用、制醋等,继续在我国国民经济发展和乡村振兴中发挥重要作用[2]。
实践证明,高粱籽粒大小的遗传改良对单产水平的提高具有重要意义。自上世纪50年代以来,我国高粱主栽品种经历了从地方品种到新品种到优良杂交种的重大更替。每一次品种的更替不仅使得单位面积产量大幅提高,而且导致选育的骨干亲本籽粒也在不断变大[3]。籽粒变大不仅能够增加产量,而且对种子活力、播后出苗、早期生长和品质性状均具有重要影响[4-6]。因此,高粱籽粒大小的研究一直是国内外高粱育种家和遗传学家关注的热点。早期研究主要从形态和生理学机制上揭示高粱籽粒大小、粒重和产量三者之间的关系,但上述研究均未与粒重性状的遗传机理结合,无法确定控制粒重的数量性状位点(quantitative trait locus,QTLs)或基因,从而限制了高粱粒重性状遗传研究进展。随着高粱分子标记技术的发展和全基因组测序的完成[7],利用连锁分析方法检测高粱粒重主效QTLs成为热门课题。大量研究报道了高粱粒重的QTLs定位[8-13],但由于受作图群体亲本遗传背景、群体类型和群体大小、标记类型差异等的影响,检测到影响粒重QTLs的数量和位置存在很大差异,且多数位点对粒重表型的贡献率较低,通过QTL克隆的方法克隆这些影响粒重性状的基因,并深入研究上述基因的功能还存在很大困难。到目前为止,通过QTLs图位克隆的方法对高粱粒重相关基因的克隆和功能研究报道较少,远远落后于水稻、拟南芥和玉米粒重性状的分子遗传研究。
基于高通量测序技术的全基因组关联分析(genome-wide association study,GWAS)方法的发展,为有效获取目标性状的候选基因提供了新途径。与传统连锁作图方法相比,GWAS方法可同时对自然群体内遗传多样性丰富的每个个体进行高通量测序,在获得数以百万计的单核苷酸多态性(single-nucleotide polymorphism, SNP)分子标记的基础上,运用不断发展的统计方法将目标性状的表型与基因型进行关联分析,从而快速准确地定位到影响目标性状的染色体区段或基因。该方法因研究材料来源广泛,可有效捕获个体间丰富的变异,节省构建遗传群体所需时间,具有更高的分辨率[14]等特点,迅速成为传统双亲种群作图的替代方法和研究复杂性状遗传的有力工具。目前GWAS已广泛应用于动植物的功能基因挖掘,并取得了显著的成绩[15]。尽管近年来利用GWAS方法解析农作物重要农艺性状的遗传机理发展迅猛,但有关高粱千粒重全基因组关联分析和候选基因的研究报道较少,仅少数研究组报道了高粱粒重性状的GWAS分析[16-20]。这些研究所采用的方法部分为基于单位点分析,或不超过两种模型,所得结果难以相互比较。基于此,本研究以242份籽粒高粱品种(系)组成的自然群体为供试群体,连续3年在杭州、贵阳、乐东和陵水4个点7个环境下测定千粒重,利用多位点GWAS分析软件中的6种模型进行全基因组关联分析,鉴定影响千粒重性状的显著性位点,挖掘、预测相关候选基因,旨在为进一步开展高粱千粒重基因的克隆及其遗传改良提供依据。
1 材料与方法
1.1 试验材料
供试材料为242份籽粒高粱种质资源,主要为我国的地方品种、育成品种及高世代育种材料,来自中国农业科学院作物科学研究所作物种质资源中心、浙江省农业科学院作物与核技术利用研究所以及贵州省旱粮研究所。
1.2 田间试验与表型调查
2018—2020年连续3年分别在贵州贵阳(18GY、19GY和20GY)、浙江杭州(18HZ和19HZ)、海南乐东(20LD)和海南陵水(20LS)共进行7次试验。小区长2.5 m,行距60 cm,株距25 cm,人工点播,适时进行间苗定苗、追肥、除草和病虫害防治等管理工作。在抽穗期,每小区选择具有代表性的3个穗子用硫酸钠纸袋套袋自交,在整株完成授粉时,将纸袋换成网袋,直至种子成熟。收获后自然晒干,再脱粒、精选和考种分析。千粒重具体测量方法:单株全株脱粒后,选择饱满无损伤的籽粒(>100粒),利用SC-G型自动考种分析及千粒重仪系统(杭州万深检测科技有限公司)进行计数和测定,重复取样3次,保证3次称重结果差异小于0.1 g(5%以内)为有效的3个数据,取平均值。利用SPSS 21.0软件对各个环境的千粒重表型进行平均值、标准差、频率分布等描述性统计分析和相关性分析,利用R 4.03软件绘图。
1.3 基因型检测
采用CTAB法[21]提取242份高粱资源叶片DNA,质检合格后利用HiSeq 4000高通量测序仪(Illumina,美国)进行测序。利用BWA软件将过滤后的测序数据比对到高粱参考基因组BTx623的序列上(https://phytozome-next.jgi.doe.gov/info/Sbicolor_v3_1_1)。利用GATK软件进行群体SNP检测,用Plink软件进行SNP过滤,筛选条件:SNP最小等位基因频率(minimum allele frequency,MAF)>5%,缺失率(missing rate, MR)<20%。最终获得2 015 850个高质量SNPs用于后续全基因组关联分析和单倍型分析。
1.4 全基因组关联分析
对2018—2020年贵阳、杭州、乐东和陵水7个环境获得的千粒重性状进行全基因组关联分析。为了提高关联分析的准确性和可靠性,本研究利用mrMLM 4.0软件[22]进行性状多位点关联分析(multi-locus GWAS methods),即同一染色体上物理距离≤20 kb 的所有显著关联SNP位点总结为1个。该软件包含mrMLM[23]、FASTmrMLM[24]、FASTmrEMMA[25]、ISIS EM-BLASSO[26]、pLARmEB[27]、pKWmEB[28]6种关联模型。K矩阵(亲缘关系矩阵)由mrMLM软件进行计算。GWAS分析时所有参数均设置为默认值。显著关联的SNPs阈值为LOD(logarithm of odds)≥3.0[26]。当显著SNP位点(物理距离不超过50 kb)满足至少在3个模型中检测到,或者在2个以上试验中检测到,即认为该SNP位点可靠,将其作为1个数量性状核苷酸位点(quantitative trait nucleotides,QTN)[29]。QTN的命名参照Linge等[29]的方法,如qtnTGW1.1表示在第1染色体上控制千粒重的第一个QTN,利用RectChr 1.31软件绘图。
1.5 候选基因预测
根据已克隆的水稻(Oryzasativa)和玉米(ZeamaysL.)粒重相关基因的蛋白序列,在Phytozome 13数据库中搜索高粱的同源基因,将搜索出来的同源基因的物理位置与本研究中检测到的千粒重显著关联的QTNs所在的物理位置进行比较,以QTNs峰值所在位点上下游50 kb范围为界,若同源基因落于该范围内,则视为可能的候选基因。
1.6 候选基因的转录本表达水平差异分析
对小粒品种654和大粒品种LTR108在穗发育的3个阶段进行取样,选取<5 cm幼穗、10~15 cm幼穗以及抽穗后5 d三个时期的穗样品,提取总RNA[13]。按照NEBNext®UltraTMRNA文库构建试剂盒(New England Biolabs,美国)操作说明构建转录组文库,质检合格后在NovaSeq 6000平台(Illumina,美国)进行测序。测序完成后,得到每个样品的原始数据(raw data),经过滤处理,通过软件Filter-FQ去除接头序列、未知序列超过10%的序列以及低质量的序列后获得清除数据(clean data),利用Hisat2软件将clean data锚定到高粱参考基因组BTx623上。基因转录组表达水平是通过FPKM(fragments per kilobase of transcript per million mapped reads)方法进行归一化处理。每时期的样品均取3次生物学重复。
1.7 候选基因的单倍型分析
基于242份高粱种质资源的重测序数据,挑选出千粒重候选基因(Sobic.002G367300、Sobic.002G367600、Sobic.003G411900、Sobic.009G024600和Sobic.010G210100)的多态性SNP位点,利用CandiHap 1.30软件对候选基因进行单倍型分析,每个候选基因选择前3种主要单倍型(≥10份材料)进行千粒重差异显著性分析。
2 结果与分析
2.1 千粒重表型统计分析
242份高粱种质资源千粒重表型在7个环境中均存在广泛的遗传变异。单个环境下测得的千粒重最大值和最小值的差值(极值)范围在23.64~31.96 g之间,其中2020年陵水的差异最小,然后依次是2018年贵阳(24.32 g)、2020年乐东(26.71 g)、2020年贵阳(27.07 g)、2019年贵阳(27.98 g)、2018年杭州(28.67 g),差异最大为2019年杭州。同一环境不同年份极值存在明显差异,2018年杭州与2019年杭州极值相差3.29 g,2019年贵阳与2018年贵阳极值相差3.66 g。单个环境千粒重平均值范围为21.87~26.89 g,变异系数范围为0.19~0.25(表1)。
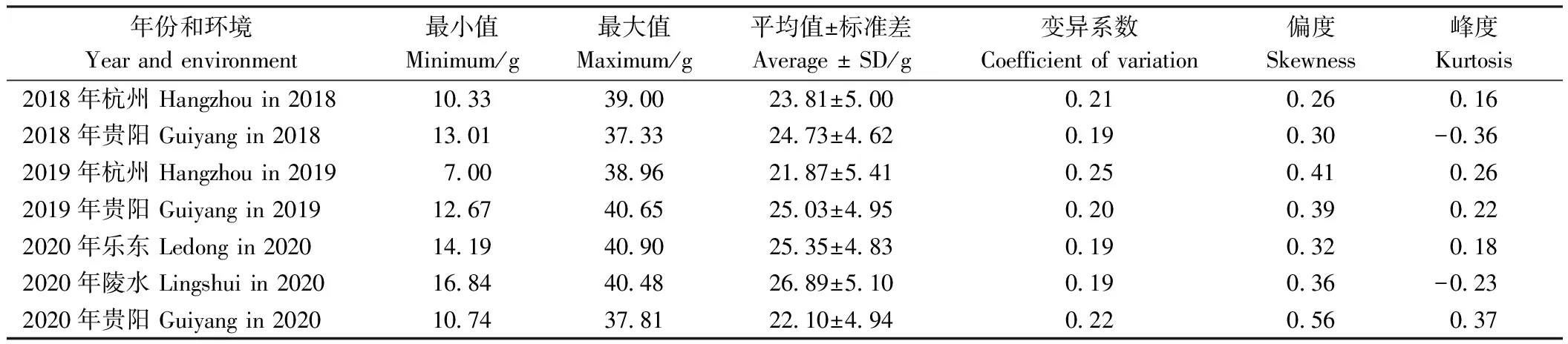
表1 3年7个环境关联群体千粒重的统计分析
从频率分布图(图1)可以看出,242份高粱种质资源的千粒重在不同年份呈现广泛的连续变异。2018年贵阳和2021年陵水的千粒重分布更趋于平展,2020年贵阳的千粒重分布具有右侧长尾。不同年份千粒重的峰度和偏度的绝对值均小于1(表1),表明千粒重值在不同年份均服从正态分布,呈现出典型的数量性状特点,受多基因控制。
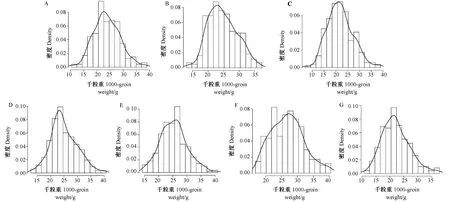
注:A:2018年杭州;B:2018年贵阳;C:2019年杭州;D:2019年贵阳;E:2020年乐东;F:2020年陵水;G:2020年贵阳。
相关系数可以反映同一品种在不同环境中表现的一致性和可重复性。2018年杭州与2019年杭州两个环境间的相关系数虽达到极显著水平,但低于0.50。2018年贵阳与2019年贵阳以及2019年贵阳与2020年贵阳间的相关系均为0.71,而2018年贵阳与2020年贵阳的相关系数为0.60;其余环境间的相关系数均界于0.52~0.69之间(图2)。表明相同地点,因年际间气象因素的差异,同一品种的表现会出现差异。

注:** 表示在P<0.01水平相关性显著。
2.2 千粒重全基因组关联分析
利用mrMLM 4.0软件包中的mrMLM、FASTmrMLM、FASTmrEMMA、ISIS EM-BLASSO、pLARmEB、pKWmEB 6种多位点关联模型对7个环境的千粒重进行全基因组关联分析,共检测到323个与千粒重显著关联的SNPs,分布在10条染色体上。LOD值范围是3.0~35.8, 可解释0.4%~26.6%的表型变异。比较不同模型检测到的显著性位点数目发现,FASTmrMLM模型检测到的最多(74个),然后依次是pKWmEB模型(66个)、pLARmEB模型(62个)、mrMLM模型(52个)、ISIS EM-BLASSO模型(42个)、FASTmrEMMA模型(27个)。有4种模型(FASTmrMLM、ISIS EM-BLASSO、pLARmEB和pKWmEB)均在2018年杭州环境点检测到最多的显著性位点,另外2种模型(mrMLM和FASTmrEMMA)则在2019年贵阳环境点检测到最多的显著性位点。有4种模型(mrMLM、FASTmrMLM、FASTmrEMMA和pKWmEB)在2019年杭州环境点检测到的显著性位点数最少,另有ISIS EM-BLASSO和pLARmEB模型分别在2020年陵水和2020年贵阳环境点检测到的显著性位点数最少。同一模型检测到的显著性位点在各条染色体上的分布不均衡。mrMLM模型在7个环境中检测到的显著性位点主要分布在第2和第5染色体上,第8染色体上的最少,仅在2020年乐东环境点检测到1个显著性位点。FASTmrMLM模型在第3染色体上检测到的显著性位点最多,第6染色体上最少,仅在2018年杭州环境点检测到1个显著性位点。FASTmrEMMA模型在第2和第6染色体上各检测到6个显著性位点,在第1、第3和第9染色体上没有检测到显著性位点。ISIS EM-BLASSO模型在第10染色体上检测到的显著性位点最多,其他染色体上分布着2~7个不等的显著性位点。pLARmEB模型在第3染色体上检测到的显著性位点最多,其他染色体上分布着1~11个不等的显著性位点。pKWmEB模型在第3和第5染色体上各检测到13个显著性位点,在第6染色体上显著性位点数最少,仅1个。
分析上述显著性位点,发现不同模型间存在重复和交叠结果,可以合并成同一个QTN,合并条件为:至少在3种模型中检测到,或者在2个以上环境中检测到相同的SNP,即视为一个可信的QTN。结果发现,共有96个显著性QTNs位点与千粒重相关联,这些位点不均匀地分布在10条染色体上,依次为6个、16个、15个、7个、16个、2个、10个、3个、7个和14个(表2、图3)。同一种模型在不同环境中均检测到的QTNs有7个,占总数的7.4%。在同一环境中,3种以上模型均检测到的QTNs有48个,占51.1%。有6个QTNs在3个环境下被检测到,单个QTN可解释1.0%~7.1%的表型变异。qtnTGW3.2和qtnTGW9.1均在2019年贵阳、2020年贵阳和2020年陵水环境点被检测到,qtnTGW3.13在2018年杭州、2020年贵阳和2020年陵水环境点被检测到。qtnTGW4.7和qtnTGW5.15在2019年贵阳、2020年贵阳和2020年乐东环境点被检测到。qtnTGW10.2在2019年杭州、2020年贵阳和2020年陵水环境点被检测到。在2个环境下检测到的QTNs有35个,占36.5%。值得注意的是,未发现一个模型在7个环境下均检测到与千粒重显著关联的共同QTNs,也未发现6个模型均检测到与千粒重显著关联的共同QTNs。
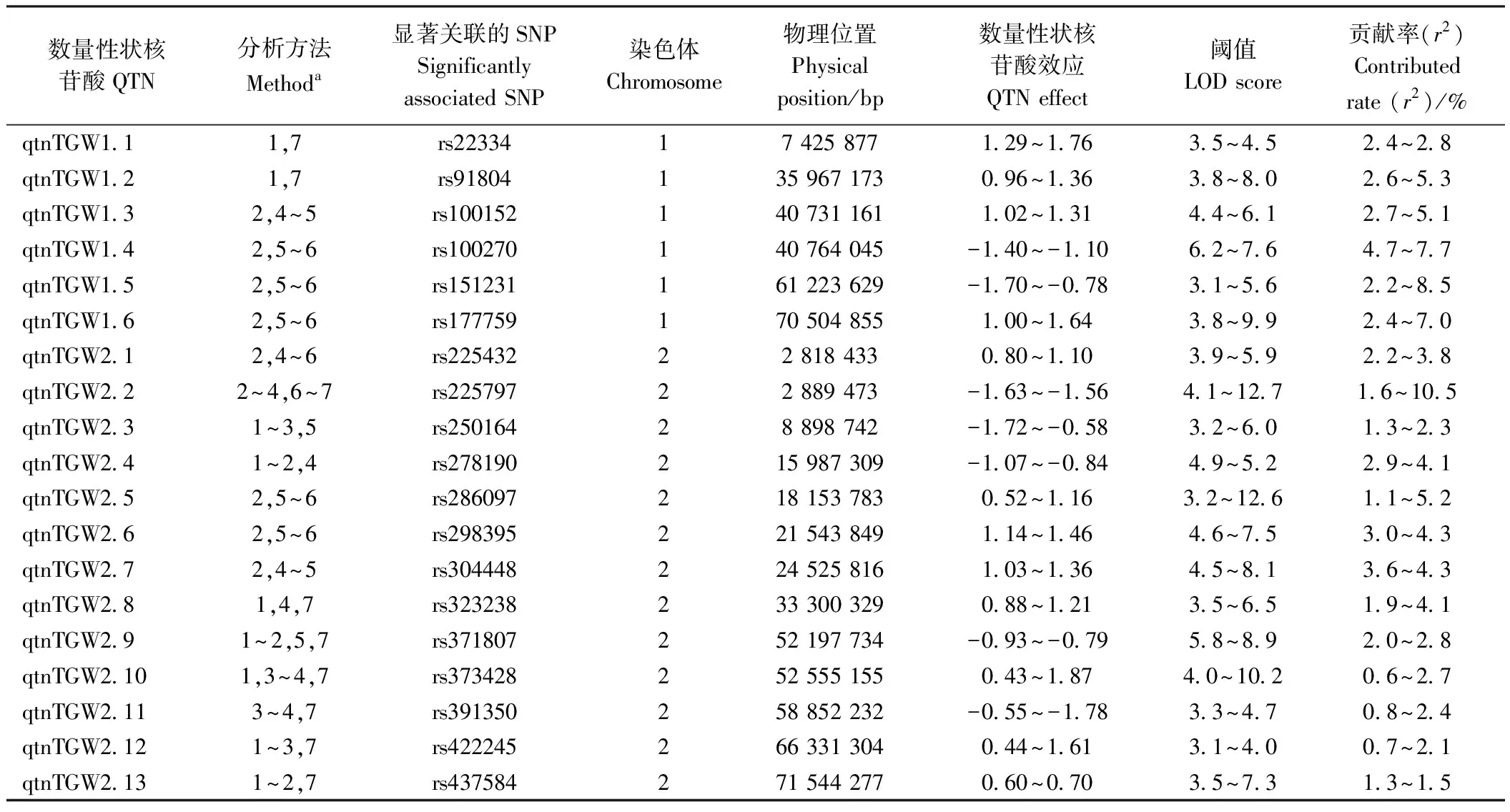
表2 利用mrMLM4.0软件中三个以上模型或两个环境中检测到与千粒重显著性关联的QTNs
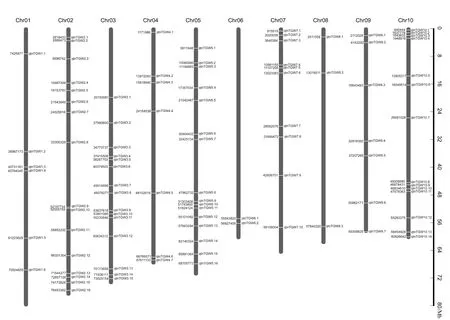
图3 影响千粒重的QTNs在染色体上的分布
2.3 千粒重候选基因分析
根据已克隆的水稻和玉米粒重相关基因的蛋白质序列,在高粱中共搜索到111个同源基因(https://www.ricedata.cn/;https://www.maizegdb.org/;https://phytozome-next.jgi.doe.gov/)。这些基因不均匀地分布在10条染色体上,其中第1染色体上数量最多,达23个,第5染色体上最少,仅1个[19]。将这些基因的物理位置与本研究中所检测到的QTNs所在区域进行比较,发现有5个与水稻粒重相关基因同源的高粱基因位于4个QTNs区间内,其他QTNs区间内均不包含已克隆的水稻粒重相关基因的同源基因。在2018年杭州和2020年陵水环境点同时检测到的千粒重qtnTGW2.15,所在区间内包含Sobic.002G367300和Sobic.002G367600基因:前者与水稻GW7/GL7同源;生物学功能显示GL7编码拟南芥LONGIFOLIA蛋白的同源蛋白,调节细胞的纵向伸长[30];GW7表达量上调,能增加谷粒的纵向细胞分裂,并减少横向细胞分裂,导致谷粒变得细长。后者与水稻BG2同源;与野生型相比,bg2-D突变体增高,同时粒长、粒宽、粒厚以及千粒重均显著增加[31]。在2018年贵阳环境点利用mrMLM、FASTmrMLM、pLARmEB、pKWmEB和ISIS EM-BLASSO 5种模型均检测到的千粒重qtnTGW3.13,所在区间内包含Sobic.003G411900基因,与水稻OsARF4同源;生物学功能显示OsARF4为生长素应答因子,与OsGSK5/OsSK41互作负向调控籽粒大小和粒重[32]。在2019年贵阳、2020年贵阳和2020年陵水环境点同时检测到的千粒重qtnTGW9.1,所在区间内包含Sobic.009G024600基因,与水稻RSR1同源;生物学功能显示RSR1是调控淀粉合成的AP2/EREBP家族的转录因子;淀粉作为水稻胚乳的主要组成成分,其含量和结构直接影响着水稻种子的品质[33]。在2018年贵阳环境点利用FASTmrMLM、pLARmEB和pKWmEB 3种模型均检测到千粒重qtnTGW10.11,所在区间内包含Sobic.010G210100基因,与水稻TGW6同源;生物学功能显示TGW6不仅直接控制胚乳的长度,而且间接参与源到库的碳水化合物的运输。在库器官(籽粒)中,日本晴的tgw6等位基因能通过控制IAA供应影响合子体到细胞化阶段的转变,从而限制细胞数目的增加和籽粒长度,而Kasalath中tgw6等位基因功能丧失会通过影响源器官的生长增加粒重,从而导致水稻产量显著增加[34]。
2.4 千粒重候选基因的转录本表达水平差异分析
图4为高粱小粒品种654和大粒品种LTR108穗发育时期5个候选基因的转录组表达量差异分析,可以看出5个候选基因的表达模式存在明显差异。其中Sobic.002G367300基因在小粒品种654三个穗发育时期表达量缓慢升高,而大粒品种LTR108在前两个时期表达量几乎没有变化,仅在抽穗后5 d表达量明显下降。在前两个时期小粒品种654的表达量低于大粒品种LTR108,而抽穗后5 d的表达量显著高于大粒品种LTR108。Sobic.002G367600基因在小粒品种654的表达量随着穗发育的完成逐渐降低,而大粒品种LTR108的表达量呈现高-低-高的趋势,且在<5 cm幼穗时期的表达量最高。大粒品种LTR108的表达量在三个时期均高于小粒品种654,且在<5 cm幼穗时期和抽穗后5 d的表达量是小粒品种654的3倍以上。Sobic.003G411900基因在两个品种中的表达量趋势呈现一致,均为随着穗发育的完成,表达量逐渐降低。三个时期小粒品种654的表达量均高于大粒品种LTR108。Sobic.009G024600基因在两个品种中表达量的趋势也呈现一致,均为前两个时期低,在抽穗后5 d表达量明显升高。Sobic.010G210100基因在两个品种中的表达量均较低(FPKM<1),该基因在小粒品种654中三个时期的表达趋势为高-低-高,而在大粒品种LTR108中的表达趋势是低-高-低。
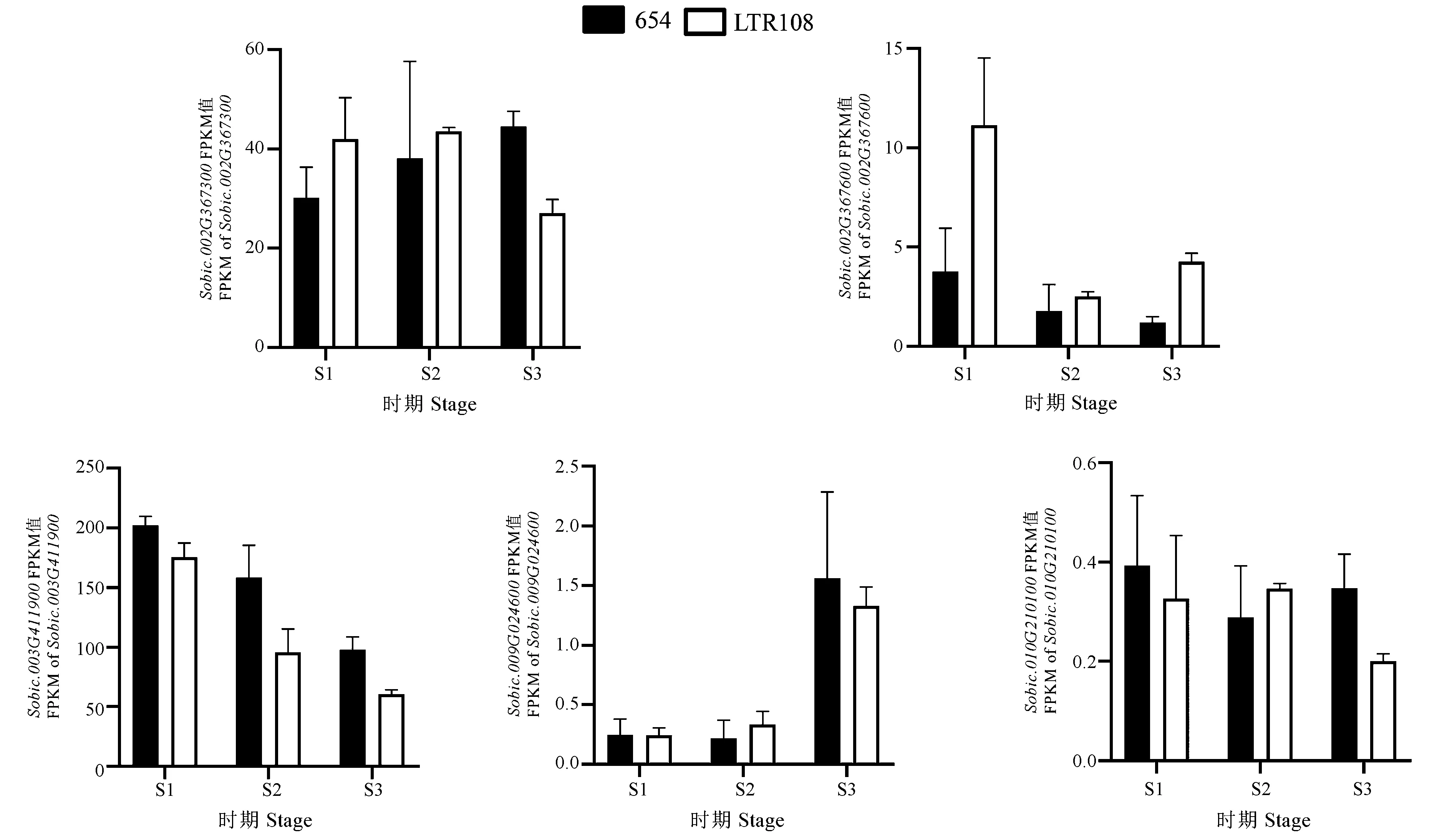
注:S1: < 5cm幼穗;S2: 10~15cm幼穗;S3: 抽穗后5 d的穗子。
2.5 候选基因单倍型分析
为了调查候选基因中变异SNP位点对表型的影响,本试验开展了Sobic.002G367300、Sobic.002G367600、Sobic.003G411900、Sobic.009G024600和Sobic.010G210100等5个基因的包含10个及以上品种的单倍型分析,如图5所示。候选基因Sobic.002G367300在第1个外显子(包含3个非同义突变和1个同义突变)、第1个内含子(包含13个多态性SNPs)、第2个外显子(包含1个同义突变)和第5个外显子(包含2个非同义突变)共存在20个多态性SNPs,这些多态性SNPs将110个籽粒高粱划分为Hap1、Hap2和Hap3,千粒重分别为24.63、23.35和25.93 g。在该候选基因中,Hap3千粒重最大,与Hap2差异达显著水平(P<0.05),为优势单倍型。候选基因Sobic.002G367600在其5′-UTR非编码区(包含1个多态性SNP)和第1个外显子(包含2个非同义突变)共存在3个多态性SNPs,这些多态性SNPs将188个籽粒高粱划分为Hap1、Hap2和Hap3三种主要单倍型,千粒重分别为23.77、24.77和25.14 g,3种主要单倍型间的差异未达到显著水平。候选基因Sobic.003G411900在第2、第6、第7、第14个内含子,第8个外显子和3′-UTR共存在6个多态性SNPs,这些多态性SNPs将98个籽粒高粱划分为Hap1、Hap2和Hap3三种主要单倍型,千粒重分别为23.61、25.44和24.75 g,3种主要单倍型间的差异也未达到显著或极显著水平。候选基因Sobic.010G210100在其5′-UTR非编码区(包含1个多态性SNP)、第1个内含子(包含2个多态性SNPs)、第3个外显子(包含1个多态性SNP)和3′-UTR(包含1个多态性SNP)共存在5个多态性SNPs,这些多态性SNPs将171个籽粒高粱划分为Hap1、Hap2和Hap3三种主要单倍型,千粒重分别为23.46、25.53和22.49 g,Hap1和Hap2之间千粒重的差异达极显著水平(P<0.01),Hap1和Hap3之间千粒重的差异不显著,Hap2和Hap3之间千粒重的差异达显著水平。候选基因Sobic.009G024600的内含子和外显子未发现差异SNP位点,故未进行单倍型分类。
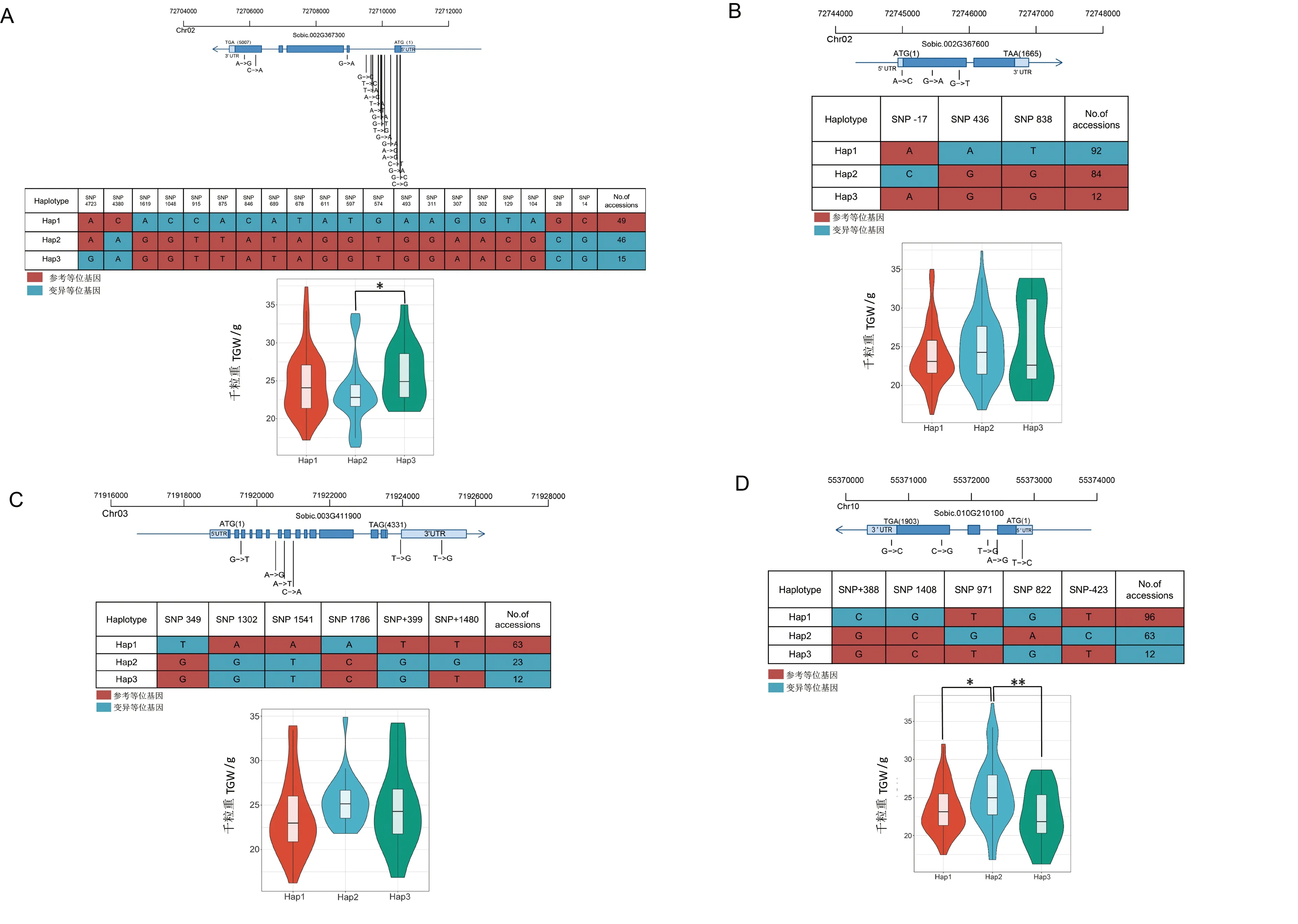
图5 候选基因单倍型分组及每种单倍型SNP 组成
3 讨论
作物粒重是典型的由微效多基因控制的数量性状,其遗传基础可以通过基于连锁作图的QTL定位和基于连锁不平衡的GWAS分析进行阐明。为提高QTL定位和GWAS分析的准确性和后续开展基因克隆的成功率,在不同遗传背景和多个环境下调查粒重性状表型,开展QTL定位和GWAS分析,鉴定稳定的主效位点是前提。为阐明高粱粒重遗传基础,本研究利用同一套高粱自然群体在4个试验点不同年份调查千粒重表型数据并开展GWAS分析。7个环境千粒重的最大值、最小值、变幅、频率分布图均存在明显差异。同一个地点不同年份之间、不同地点相同年份之间,以及在不同气候类型(亚热带湿润温和型气候的贵阳、亚热带季风气候的杭州以及热带季风气候的陵水和乐东)条件下的表现都不完全一致。GWAS分析结果同样反映了这种差异:7个环境下未检测到相同的QTN,6个QTNs在3个环境下检测到,2个环境下检测到的QTNs有35个,在单一环境下检测到的QTNs占多数,表明高粱粒重的遗传基础非常复杂。本研究检测到的影响千粒重的QTNs,为探究高粱千粒重的遗传提供了有用信息。
GWAS是基于连锁不平衡来识别分子标记之间或候选基因与性状之间关系的方法。如果两个基因座位间的不平衡是来源于群体结构或亲缘关系,则会影响GWAS结果的准确性。此外,GWAS研究中仅能检测到一小部分变异,对于由稀有变异、结构变异、上位性互作及基因与环境互作等因素造成的遗传变异难以检测到[35]。为了提高GWAS分析的准确性,需进一步完善统计分析方法。基于混合线性模型(mixed linear model,MLM)的GWAS因兼顾固定效应和随机效应,显著提高了数量性状位点检测的功效,被视为标准方法[36]。然而,MLM是单位点模型的分析方法,一次只能进行单个位点的检测,与受多基因控制的复杂性状的真实遗传模型不符[37]。随着GWAS模型的不断发展,同时兼顾所有位点信息的多位点GWAS方法得以开发[23]。与单位点全基因组关联分析模型相比,多位点SNP随机效应混合线性模型更加接近真实的动植物遗传模型[38],即使应用了较为宽松的关联阈值,也具有更高的统计功效和更低的假阳性率[39]。本研究在高粱中首次利用包含mrMLM、FASTmrMLM、FASTmrEMMA、pLARmEB、pKWmEB、ISIS EM-BLASSO 6种多位点模型的mrMLM 4.0软件检测影响千粒重的QTN,共检测到323个显著性位点。不同模型检测到的显著性位点存在差异,本研究中FASTmrEMMA模型检测到显著关联位点最少,FASTmrMLM模型检测到的最多,在开展GWAS分析时,选择不同的统计分析方法会影响检测效果,因此,进行多种统计分析方法的比较和甄选,将有助于提高GWAS分析的准确性。本研究中最终合并和整合到96个影响千粒重的QTNs,远远多于以前单个研究报道的高粱粒重GWAS分析的数量,检测效率大大提高。
到目前为止,高粱中仅少数文献报道了粒重GWAS分析结果。其中Upadhyaya等[16]利用39个微卫星标记(simple sequence repeat,SSR),在第10染色体的50 197 002-51 527 907 bp区间内检测到一个影响粒重的主效QTL;Sakhi等[17]利用98个SSR标记,对107个高粱种质采用MLM中的条件型Q+K模型在第8染色体的57 579 092-58 285 925 bp检测到一个影响百粒重的主效QTL。两个研究因标记数量少,基因组上的覆盖度低,检测到的QTL数量极其有限。Boyles等[18]利用268 830个SNPs、390个品种组成的关联群体以及混合线性模型(MLM)和贝叶斯稀疏线性混合模型(Bayesian sparse linear mixed model,BSLMM)对千粒重性状开展GWAS分析,两年环境共检测到19个显著关联的SNPs。Tao等[19-20]利用同一套材料对千粒重开展GWAS分析,在2020年的报道中利用FarmCPU模型在自然群体中检测到58个影响千粒重的显著关联位点,巢式关联群体(backcross-nested association mapping,BC-NAM)群体中检测到59个影响千粒重的显著性关联位点,有42个位点是两个群体中同时被检测到的;2021年的报道中利用始花时去除半个穗子处理的方法对千粒重开展GWAS分析,在自然群体中检测到79个影响千粒重的显著关联位点,BC-NAM群体中检测到69个影响千粒重的显著性关联位点,有57个位点是两个群体中同时检测到的。去掉半个穗子和保留完整穗子检测到的共同显著性位点有18个。将本研究与上述5个研究结果进行比较,发现所检测到的众多显著性位点中,仅qtnTGW2.12、qtnTGW2.13、qtnTGW3.13、qtnTGW4.6分别与Tao等[19-20]检测到的qGS2.5、qGS2.8、qGS3.7和qFHGS4.11重叠。推测可能由于GWAS采用的分析群体不同,本研究分析群体主要由中国高粱种质组成,而上述关联群体所涉及的品种主要来源于非洲和美国的高粱。
水稻、玉米和高粱同属于禾本科二倍体作物。在漫长的驯化过程中,一些驯化性状(如果实增大、种子不易落粒、休眠被打破、生育期同步等)被人们有意或无意选择并保存下来,而涉及这些相似性状的基因因存在同源性和共线性,也被很好地保存下来,因而可能存在功能保守的基因组区域。水稻中克隆的粒重GS3在玉米和高粱中的同源基因均被证实影响着籽粒大小和粒重[13,40]。本研究检测到4个千粒重QTNs,其所在区间内含5个水稻中已经克隆的粒重同源基因,这些基因在小粒品种654和大粒品种LTR108穗发育的三个时期表达量存在差异,有可能调控高粱千粒重,但需要进一步验证相关基因的功能。
4 结论
本研究利用多位点全基因组关联分析方法中的6种模型对高粱千粒重性状进行了全基因组关联分析,在7个环境下共检测到96个与千粒重显著关联的QTNs位点,其中6个位点在3个环境中被同时检测到,另外35个在2个环境中被同时检测到。有5个与水稻粒重相关基因同源的高粱基因位于在4个QTNs区间内,这些基因可能是调控籽粒发育的候选基因。
