多靶点CAR-T治疗血液系统恶性肿瘤研究进展
2022-10-24冯友琴张棋琦胡永仙黄河
冯友琴 张棋琦 胡永仙 黄河
嵌合抗原受体T细胞(CAR-T)治疗复发难治性血液系统恶性肿瘤疗效显著,成为近年来肿瘤治疗领域最重大的突破之一。据报道CD19 CAR-T治疗复发难治性急性B淋巴细胞白血病(B-ALL)及复发难治性B细胞非霍奇金淋巴瘤(B-NHL)的完全缓解率(CRR)分别是60%~90%[1-3]和40%~50%[4-5];B细胞成熟抗原(BCMA)CAR-T治疗复发难治性多发性骨髓瘤(RRMM)的CRR为39%~80%[6-8];G蛋白偶联受体C5家族亚型D蛋白(GPRC5D)CAR-T(OriCAR-017)治疗RRMM的CRR/严格意义的CRR(sCRR)为60%(NCT05016778);CD7 CAR-T治疗复发难治性T细胞急性淋巴细胞白血病(T-ALL)或淋巴母细胞淋巴瘤(LBL)的CRR可达87.5%~94.0%[9]。尽管单靶点CAR-T疗效显著,但仍有部分复发难治性血液病患者存在治疗无效或治疗后复发。据文献报道,复发难治性B-ALL患者在使用CD19 CAR-T治疗后的复发率约为17%~57%,其中CD19阴性复发率为7%~25%[1,10]。
CAR-T治疗后复发是目前临床面临的巨大挑战,其涉及机制主要包括抗原依赖性因素(抗原逃逸、抗原脱落或抗CAR抗体)及T细胞驱动性因素(CAR-T耗竭、免疫抑制性肿瘤微环境)[11]。在血液系统肿瘤研究领域,靶抗原的缺失是最具特征的耐药机制,靶抗原阴性克隆的复发具有多种潜在机制,如移码/错义突变或可变剪接、预先存在的抗原阴性亚克隆选择性扩增、表位遮蔽和谱系转换/转分化等[12]。另有研究表明CAR-T还可通过胞啃作用将肿瘤细胞表面的靶抗原转移到自身表面,降低肿瘤细胞上靶点的密度,并引起CAR-T之间相互攻击,最终导致T细胞的耗竭与活性降低[13]。与单靶点CAR-T相比,多靶点CAR-T可降低抗原丢失的可能性,并可增加其抗肿瘤活性[13-14]。目前双靶点CAR-T研发及临床转化日渐增多,从报道的综合临床数据可看出,双靶点CAR-T展现出极富前景的应用价值。本综述总结了目前应用多靶点CAR-T治疗复发难治性血液系统恶性肿瘤临床转化研究的最新进展。
一、多靶点CAR-T结构
用于构建多靶点CAR-T的CAR结构主要包括:鸡尾酒/序贯CAR、共转导型CAR、并联型CAR、二价串联型CAR、二价环形CAR。
1.鸡尾酒/序贯CAR(图1A):分别用两种不同病毒载体转导的T细胞可产生两种单独的CAR-T产品,然后将两种单独的CAR-T以1∶1的比例混合在一起,同时或序贯输注[15-16]。该方式可灵活调整组合和剂量,但生产成本高,从临床实践观察,两个CAR-T共同或序贯回输,可能以某一靶点CAR起主要作用。
2.共转导型CAR(图1B):用两个病毒载体同时共转导,每个载体编码一个单独的CAR结构。其最终的混合产物中包含两个单独的单靶点CAR-T和一个双靶点CAR-T[16]。但细胞产品中会存在没有转上CAR的T细胞,质控比较困难。
3.并联型CAR(图1C):即双顺反子CAR,带有不同单链抗体片段(scFv)的CAR分子转入同一个T细胞,其中每个CAR靶向不同的抗原结合域,且具有各自独立的信号传导途径[16-18]。但CAR分子较大易导致病毒包装或转导效率出现问题,且两个胞内信号更易导致CAR-T耗竭。
4.二价环形或串联型CAR(图1D、1E):将一个二价病毒载体引入T细胞而产生双结构域。根据scFv的轻链可变结构域(VL)和重链可变结构域(VH)排列顺序的不同,可分为两种结构:串联和环形CAR。两种结构的胞内均只有一个共同的信号传导途径[18-19]。串联结构是由一个scFv的VL-VH直接连接另一个scFv的VL-VH,而环状结构则由一个scFv的VL-VH夹在另一个scFv的VH-VL中间形成。该类型CAR-T胞外结构复杂,质粒构建难度大。
二、多靶点CAR-T临床研究进展
截至目前,多靶点CAR-T治疗复发难治性血液病的临床研究已取得重大进展。同时靶向CD19/CD20、CD19/CD22、CD19/BCMA或BCMA/CS1的双靶点CAR-T在通过临床前研究证实具有强大的抗肿瘤活性后已进入临床试验阶段。本部分举例综述在不同血液系统肿瘤中多靶点CAR-T临床试验的安全性与疗效。见表1。
1.复发难治性恶性B细胞血液系统肿瘤:多项CD19/CD20及CD19/CD22双靶点CAR-T临床试验公布的结果表明,二者治疗复发难治性B-NHL、复发难治性B-ALL均具有较高的缓解率且不良反应可控,同时也是目前开展最多的双靶点CAR-T治疗临床试验。韩卫东教授团队在2017年5月发起了一项开放标签、单臂Ⅰ/Ⅱ期临床试验(NCT03097770),以评估CD19/CD20双靶点CAR-T(TanCAR7-T)治疗B-NHL的疗效及安全性[19]。截至2021年3月,研究共入组87例患者,总缓解率(ORR)达78%,CRR可达70%。9例患者发生3~4级CRS,仅2例患者发生3级神经系统反应(癫痫)且不良反应可控。中位随访27.7个月,仍有60%的患者维持持续缓解。
早在2020年4月韩卫东教授团队率先证实了CD19/CD22的双特异性CAR-T治疗B-ALL的安全性和有效性[20]。我中心团队近期开展了一项开放标签、剂量递增的Ⅰ期临床试验(NCT04227015),以评估CRISPR/Cas9基因编辑的通用型CD19/CD22双靶点CAR-T(CTA101)治疗复发难治性急性淋巴细胞白血病(ALL)的疗效和安全性。截至2020年8月,研究共入组6例患者,输注后的CRR为83.3%,中位随访4.3个月,获得CR或CR伴不完全血液学缓解(CRi)的5例患者中有3例微小残留病灶(MRD)持续阴性[21]。
2.复发难治性恶性浆细胞血液系统肿瘤:Ide-cel是第一个被美国食品药品监督管理局(FDA)批准用于治疗RRMM的BCMA CAR-T产品,其疗效显著、不良反应可控[6]。但单靶点BCMA CAR-T治疗后仍有约45%患者出现肿瘤复发[22],其中4%~33%CAR-T治疗后复发的患者出现BCMA抗原的丢失或下调[6,23]。同时研究发现其他浆细胞标志物和潜在的免疫治疗靶标,如CD38和CS1(SLAMF7)在疾病进展过程中仍稳定表达[24]。这些现象均表明靶向多种浆细胞抗原的CAR-T疗法可能是避免抗原逃逸的一种可行方法。
基于该现象胡豫教授团队设计了BCMA/CD38双靶点CAR-T,在体内外实验中评估其治疗的有效性和安全性后,发起一项BCMA/CD38 CAR-T治疗RRMM临床试验(ChiCTR1800018143)。该研究共入组23例患者,中位随访9个月,12例(52%)患者达sCR,另有4例(17%)获得非常好的部分缓解(VGPR)及4例(17%)出现部分缓解(PR)。87%的患者出现CRS,≥3级CRS仅22%,未发生任何级别神经不良反应。此外,该研究还发现BM38 CAR-T在9个月、12个月时可分别在77.8%、62.2%的患者体内检测得到,表明其在体内持久性较好[17]。
2022年第27界欧洲血液学年会(EHA)公布了GC012F的临床试验数据,这是一款可由亘喜生物FasTCAR平台开发的靶向BCMA/CD19双靶点自体CAR-T。该研究共入组28例复发难治性B-NHL患者,中位随访6.3个月,不同剂量水平(DL)的ORR分别为100.0%(DL1)、80.0%(DL2)及93.8%(DL3),75%患者获得sCR。3级以上CRS只有7.1%,且所有患者均未发生任何级别神经不良反应(NCT04236011;NCT04182581)。此外第27届EHA还公布了BCMA/CS1双靶点CAR-T临床试验结果,共入组13例RRMM患者,其ORR和CRR分别为76.9%和30.8%。31%的患者发生CRS,未见神经不良反应(NCT04662099)。
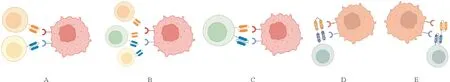
图1 多靶点CAR-T结构(A:鸡尾酒/序贯CAR;B:共转导型CAR;C:并联型CAR;D:二价环形CAR;E:二价串联型CAR)

表1 多靶点CAR-T治疗的临床试验
3.多靶点CAR-T治疗其他复发难治性恶性血液系统肿瘤:相比B细胞血液系统恶性肿瘤,双靶点CAR-T治疗复发难治性急性髓系、T细胞性血液系统恶性肿瘤面临更多挑战。
CAR-T治疗复发难治性急性髓系白血病(AML)的主要障碍在于AML细胞异质性较大、缺少高度特异性靶点。AML细胞上各种表面抗原如CD123、CD34、CD33等,同时也在正常的造血干细胞、髓系和(或)淋巴祖细胞上表达[25],针对这些靶点的CAR-T治疗相关不良反应较大。此外有研究发现AML细胞可分泌抑制T细胞增殖的免疫抑制因子,影响CAR-T在体内的扩增[26]。CAR-T治疗T-ALL或T细胞非霍奇金淋巴瘤(T-NHL)面临的亟需解决的问题主要有CAR-T的自相残杀、患者T细胞缺乏、T细胞性再生障碍性贫血及制备的自体CAR-T易被恶性T细胞污染等问题[27]。CAR-T治疗急性髓系、T细胞血液系统肿瘤有待寻找合适的靶点及改进制备工艺。
国内一款靶向CLL1/CD33的CAR-T在治疗复发难治性AML中显示出积极疗效。西部战区总医院刘芳教授团队率先证实CLL1/CD33双靶点CAR-T在小鼠体内具有强大的抗肿瘤活性,随后于2020年EHA上公布了该双靶点CAR-T的Ⅰ期临床研究数据,入组的9例复发难治性AML患者中7例达到CR,88.8%患者发生CRS但不良反应可控(NCT03795779)。其余针对复发难治性AML治疗的双靶点如CD123/CLL1(NCT03631576)、CD123/CD33(NCT04156256)也已显示出初步的疗效和安全性,相关数据尚未证实发布。
三、多靶点CAR-T临床前研究进展
虽然多靶点CAR-T治疗复发难治性血液病取得了较大进展,但同样面临着相应的临床挑战,如脱靶效应、制备困难等。越来越多的研究者致力于新型多靶点CAR-T的研发,包括BAFF CAR-T治疗RRMM、CRISPR/Cas9基因敲除CD5/CD7 CAR-T治疗T-ALL及三靶点CAR-T治疗复发难治性血液病等。同时越来越多的研究通过优化多靶点CAR-T的结构设计以减少不良反应的发生。
1.BAFF CAR-T/APRIL CAR-T:B细胞激活因子(BAFF,BlyS)和增殖诱导配体(APRIL)是肿瘤坏死因子(TNF)超家族的两个同源成员,BAFF可与表达在成熟B细胞上的3个受体BAFF-R、BCMA和穿膜蛋白活化物(TACI)结合,ARPIL可与BCMA、TACI结合,启动下游信号转导以促进恶性浆细胞的增殖与存活[28]。以BAFF或APRIL作为CAR-T胞外识别结构域,相当于可同时靶向肿瘤的多个抗原,在治疗多种B细胞肿瘤时可起协同作用。BAFF CAR-T尚处于临床前研究阶段,Wong及其研究团队[29]设计了基于BAFF配体的CAR构建体,利用非病毒TcBuster转座子系统将BAFF CAR整合到T细胞中,体内外实验均证实BAFF CAR-T具有显著的抗肿瘤能力。我中心开展的APRIL CAR-T治疗RRMM研究已转化临床(NCT04657861),相关数据尚未正式公布。
2.双特异性及分离型(Biss)CAR-T:2020年He等[30]构建出BissCAR,主要由P2A(linker)连接纳米抗体Nb157、CD3ζ和anti-TIM3-scFv、CD28、4-1BB。P2A自剪切后BissCAR最终会分裂成两个CAR。Nb157是由作者开发的序列肿瘤选择抗体和抗原检索(STAR)技术筛选出来的纳米抗体,能特异性识别在AML细胞上高表达的CD13。为了成功靶向耐药AML干细胞而不“误杀”正常骨髓造血干细胞,研究者选择了T细胞免疫球蛋白黏蛋白分子3(TIM3,在骨髓造血干细胞低表达或不表达)作为另一种共刺激信号的靶抗原。实验证实BissCAR-T未攻击K562-TIM3(CD13-TIM3+)系细胞,对NB4(CD13+TIM3-)系细胞仅表现出很低的杀伤作用,而对NB4-TIM3(CD13+TIM3+)系细胞却表现出最大的细胞毒性[30]。同时靶向CD13和TIM3的BissCAR-T可根除AML细胞,且对正常造血干细胞影响较小,具有更高的临床安全性。
3.synNotch “AND-gate” CAR-T:2017年Morsut等[31]在《Cell》上发布了一项研究,首次设计了一种基于Notch的组装式受体即synNotch受体。随后该团队利用这一平台设计了synNotch “AND-gate” CAR系统,其识别抗原A的人工受体synNotch基因,包含了抗原A的识别片段及启动抗原B识别CAR基因转录的转录因子。装载synNotch系统的CAR-T只能识别和杀伤同时表达抗原A和B的肿瘤细胞[32]。该策略为CAR-T的精准识别和治疗提供了新模式。
4.抑制型CAR-T:当某种抗原在肿瘤细胞和正常细胞上同时表达时,CAR-T治疗可能会引起脱靶效应,产生相应的不良反应。早在2013年就有研究者开发了一种抑制型CAR-T(iCAR),在CAR-T中同时表达CAR和iCAR。iCAR是经过改造的抑制性受体[如程序性死亡受体1(PD-1)、细胞毒性T淋巴细胞相关抗原4(CTLA-4)],能够识别正常组织呈递的抗原。这种CAR-T作用于正常细胞时活性会被抑制,从而精确靶向肿瘤细胞,发挥杀伤的作用[33]。此外,有些抗原在正常细胞中表达而在肿瘤细胞中不表达,利用这一特性,synNotch “OFF-gate” CAR-T在作用于正常细胞时可实现将synNotch通路激活的基因变成促凋亡相关基因,从而使T细胞发生凋亡,避免细胞治疗中交叉反应而导致的不良反应[34]。抑制型CAR-T及synNotch “OFF-gate” CAR-T均为解决脱靶效应提供了新的策略。
5.CRISPR/Cas9基因编辑CAR-T:正如上述提及,CAR-T疗法对于复发难治性T细胞血液系统肿瘤效果欠佳。针对CAR-T自杀伤这一问题,近期有研究在双特异性CAR的慢病毒转导之前进行基于CRISPR/Cas9的CD5和CD7基因敲除,功能实验证实基因敲除对细胞无明显影响。且随后的体内外实验证实相较于并联型CAR-T,串联型CD5/CD7 CAR-T具有更高的杀伤活性,且能够更有效地防止肿瘤逃逸[35]。基于CRISPR/Cas9基因编辑的CD5/CD7双靶点CAR-T研究结果为T细胞血液系统肿瘤的免疫治疗提供了新的方向。
6.三靶点CAR-T:在双靶点CAR-T疗法取得积极成果的同时,三靶点CAR-T的临床前研究也取得相应进展。在一项新的研究中,Lentigen研究人员开发出三特异性duoCAR-T ,由靶向CD19和CD20的串联CAR通过P2A自裂解肽连接到另一种靶向CD22的CAR上组成,且证实最有效的duoCAR结构包含ICOS、OX40或CD27共刺激域,而不是CD28或4-1BB。体内外实验证实duoCAR-T拥有较高的抗肿瘤效力和持久性,同时克服了肿瘤异质性和肿瘤抗原逃逸的临床挑战[36]。三靶点CAR-T疗法无疑是在双靶点CAR-T的基础上进一步降低了因抗原逃逸或丢失导致的复发风险,深度消除肿瘤细胞。但目前该技术还处于临床前研发阶段,仍需大量研究以评估其临床前毒性、产品开发工艺与质控、适应证人群、给药剂量等问题。
四、总结与展望
虽然CAR-T治疗在复发难治性血液系统恶性肿瘤中取得了卓越的疗效,但CAR-T治疗后复发的问题依然严峻。与单靶点CAR-T相比,多靶点CAR-T主要的优势在于可减少肿瘤细胞的抗原逃逸。双靶点CAR-T治疗的疗效已在部分临床试验中得到了验证,同时各个临床试验报道的严重CRS和ICANS等不良反应的发生率也较低。多靶点CAR-T可能成为未来细胞免疫治疗领域研究的趋势。
尽管如此,多靶点CAR-T治疗仍面临着前所未有的挑战。首当其冲的是靶点的选择与验证。B细胞血液系统肿瘤潜在免疫治疗靶点众多,如何优化靶点选择以更好提高疗效,还需更多基础研究与临床研究相结合。对于髓系血液系统肿瘤中双靶点的合理选择,如靶向CD33/TIM3或CLL1/TIM3仍待进一步探究。另一个亟需解决的问题是多靶点结构的优化。正如上述提到串联型双靶点CAR-T的疗效优于并联型,但串联型CAR-T开发难度较大,linker的选择也是需要关注的问题。最后就是新型CAR-T的技术优化与产品开发。自体CAR-T面临较多缺陷,如难以采集足够多的外周血T细胞、自体CAR-T不能大规模生产及从采集到制备耗时较长等问题,通用型多靶点CAR-T和诱导多能干细胞(iPSCs)来源的多靶点CAR-T为细胞治疗领域开辟了新的治疗方向。
