盐酸小檗碱对大鼠体内低剂量阿托伐他汀的药动学影响 及机制研究
2022-10-19余瑞莲廖辉雄陈海珍
余瑞莲,廖辉雄,陈海珍
(1.深圳市龙华区人民医院药学部;2.深圳市龙华区人民医院康复科;3.深圳市龙华区人民医院壹城中心社康中心药房,广东深圳 518109)
目前,心血管疾病是导致人类死亡的主要原因,而动脉粥样硬化是导致冠心病和卒中的主要原因,高胆固醇血症是一般动脉粥样硬化,特别是冠心病发展的关键因素[1]。在个体水平上,随着总血清胆固醇或低密度脂蛋白胆固醇水平的增加,动脉粥样硬化过程和早发性冠心病进展的风险逐渐增加。阿托伐他汀适用于治疗血脂异常并具有良好的安全性,其可通过抑制3-羟基-3-甲基戊二酰辅酶A还原酶的活性,有效阻断胆固醇生物合成的早期限速步骤,在极低密度脂蛋白水平升高的情况下,阿托伐他汀会降低三酰甘油水平[2]。小檗碱是一种从传统中药材中分离出来的植物性生物碱,对心血管具有保护作用[3]。临床研究表明,小檗碱可降低血浆胆固醇和葡萄糖水平,它已被用作抗炎剂、抗菌剂、抗高血压剂、抗心律失常剂、抗肿瘤剂和抗糖尿病剂[4-6]。目前,临床常使用小檗碱与他汀类药物联合治疗高脂血症[7-8]。基于此,本研究旨在探讨盐酸小檗碱对大鼠体内低剂量阿托伐他汀的药动学影响及机制,为临床应用小檗碱与他汀类药物提供指导。
1 资料与方法
1.1 试验动物一般资料60只SPF级雄性SD大鼠[周 龄10~12周,体 质 量(230±20)g]均 获自北京晶来华科生物技术有限公司[动物批号:110324220102956362;许 可 证 号:SYXK(京)2022-0015]。动物房饲养环境为恒温恒湿的屏障环境。将大鼠安置在公用笼子中,笼子放置在(21±2)°C的常规动物房间中,给予12 h光照/12 h黑暗循环,并提供食物和饮用水。本研究根据深圳市龙华区人民医院动物护理和使用委员会批准的方案(医学伦理审批号:20220112-017)进行。
1.2 试验给药和分组根据研究方案将大鼠分为以下3组:A组(n=20)大鼠首天单独给予10 mg/kg 剂量的阿托伐他汀灌胃7 d;B组(n=20)大鼠首天先给予20 mg/kg剂量的小檗碱灌胃,5 min后再给予10 mg/kg剂量的阿托伐他汀灌胃7 d;C组(n=20)大鼠给予20 mg/kg剂量的小檗碱连续灌胃6 d,在第7天给药小檗碱5 min后给予10 mg/kg剂量的阿托伐他汀灌胃。于阿托伐他汀给药前及给药后30 min、2 h、4 h、6 h、8 h、10 h、12 h通过尾静脉出血收集血样,将血样在4 °C下以2 880 r/min 离心10 min,收集上清液以获得血浆样本,将其保存在-80 °C直至使用。
1.3 试验方法
1.3.1 药物代谢动力学分析本研究使用Phoenix WinNonlin 8.1版(普林斯敦美国)药动学软件,以线性向上对数向下梯形法进行非隔室药代动力学分析。评估阿托伐他汀的血浆参数,包括达到最大血浆浓度的时间(Tmax)、最大(峰值)血浆浓度(Cmax)、终末半衰期(T1/2)、从0到无穷大的血浆浓度-时间曲线下面积[AUC(0-∞)]、从0到时间t的AUC[AUC(0-t)]、清除率(CLz/F)、平均滞留时间[MRT(0-t)和MRT(0-∞)]。通过盐酸小檗碱获得的值来计算Cmax和AUC比率。
1.3.2 精密度和准确度取阿托伐他汀的标准储备液用乙腈分别稀释成浓度为15、5、1.5 μg/mL的阿托伐他汀溶液。分别取以上各浓度的阿托伐他汀溶液各10 μL,加入100 μL的空白血浆,分别配成阿托伐他汀终浓度为100、350、1 000 ng/mL的质控样品溶液。评估样品稀释效果,以确保在用空白基质稀释后可以正确测定超出校准范围浓度上限的分析物浓度。精密度和准确度的验收标准分别为变异系数(Coefficient of Variation)低于或等于15%和偏差在±15%以内。
1.4 统计学分析本研究中的数据全部采用SPSS 20.0统计分析软件进行处理。计量资料采用(±s )表示;在重复测量资料分析前,对球性条件进行验证(Mauchly球性检验),即任意两个时间点之间的差值的方差差异无统计学意义,球性条件得到满足后可采用普通的单因素方差分析,P<0.05表示差异有统计学意义。
2 结果
2.1 血浆中阿托伐他汀精确度、准确度分析阿托伐他汀的3个质控浓度100、350和 1 000 ng/mL的精密度和准确度见表1,结果表明本方法的精密度和准确度满足要求。
表1 血浆中阿托伐他汀精确度、准确度分析(±s )

表1 血浆中阿托伐他汀精确度、准确度分析(±s )
质控浓度(ng/mL) 日内 日间测定浓度(ng/mL) 准确度(%)精密度(%) 测定浓度(ng/mL) 准确度(%)精密度(%)100 134.59±11.24 108.35 13.40 125.33±8.42 98.34 12.34 350 495.38±22.18 102.44 9.95 503.26±18.34 105.35 7.12 1 000 1 386.24±37.45 105.49 5.36 1 425.33±45.51 108.26 9.23
2.2 大鼠阿托伐他汀的药动学参数分析通过药动学软件对阿托伐他汀进行药动学参数的计算,A组、B组和C组大鼠阿托伐他汀在体内药代动力学参数的比较,差异无统计学意义(P>0.05)。见表2。
表2 大鼠阿托伐他汀的药动学参数分析(±s )
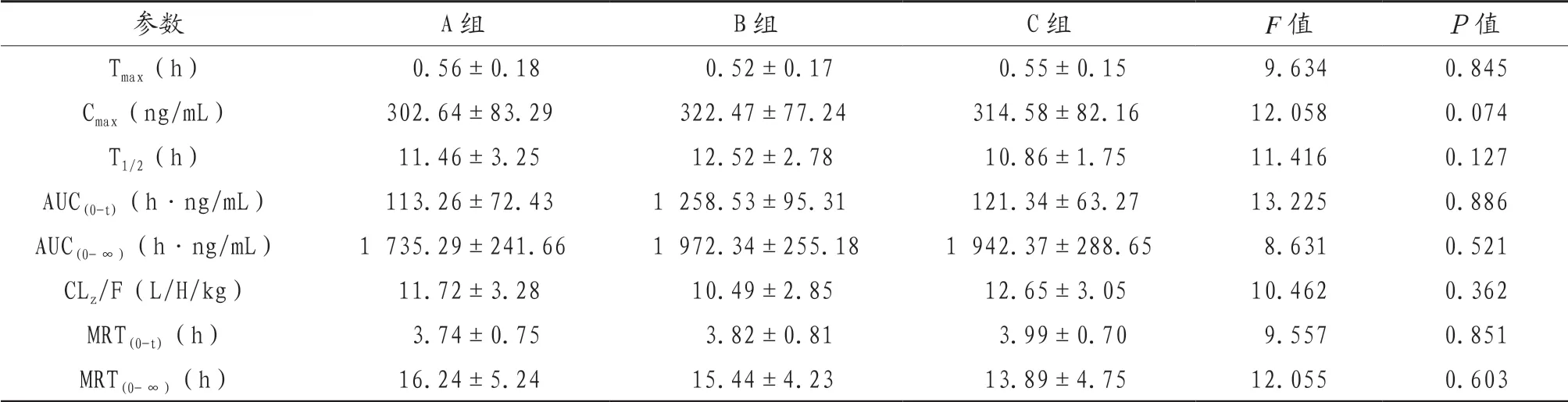
表2 大鼠阿托伐他汀的药动学参数分析(±s )
Tmax:达到最大血浆浓度的时间;Cmax:最大(峰值)血浆浓度;T1/2:终末半衰期;AUC(0-∞):从0到无穷大的血浆浓度-时间曲线下面积;AUC(0-t):从0到时间t的AUC;CLz/F:清除率;MRT:平均滞留时间,即MRT(0-t)和MRT(0-∞)。
参数 A组 B组 C组 F值 P值Tmax(h) 0.56±0.18 0.52±0.17 0.55±0.15 9.634 0.845 Cmax(ng/mL) 302.64±83.29 322.47±77.24 314.58±82.16 12.058 0.074 T1/2(h) 11.46±3.25 12.52±2.78 10.86±1.75 11.416 0.127 AUC(0-t)(h·ng/mL) 113.26±72.43 1 258.53±95.31 121.34±63.27 13.225 0.886 AUC(0-∞)(h·ng/mL) 1 735.29±241.66 1 972.34±255.18 1 942.37±288.65 8.631 0.521 CLz/F(L/H/kg) 11.72±3.28 10.49±2.85 12.65±3.05 10.462 0.362 MRT(0-t)(h) 3.74±0.75 3.82±0.81 3.99±0.70 9.557 0.851 MRT(0-∞)(h) 16.24±5.24 15.44±4.23 13.89±4.75 12.055 0.603
2.3 大鼠体内阿托伐他汀浓度分析分析不同时间大鼠体内阿托伐他汀的浓度,在给药后第30 min 、2 h、4 h、6 h、8 h、10 h和12 h,A组、B组和C组大鼠阿托伐他汀血药浓度比较,差异无统计学意义(P>0.05),见表3。
表3 大鼠体内阿托伐他汀浓度分析(ng/mL,±s )

表3 大鼠体内阿托伐他汀浓度分析(ng/mL,±s )
注:在重复测量资料分析前,对球性条件进行验证(Mauchly球性检验),即任意两个时间点之间的差值的方差差异应无统计学意义,球性条件得到满足后可采用普通的单因素方差分析。
时间 A组 B组 C组 F值 P值30 min 246.18±24.52 265.37±28.24 255.19±26.43 11.625 0.523 2 h 185.35±26.38 196.62±24.33 211.35±28.37 9.775 0.083 4 h 117.36±18.49 123.57±17.43 115.34±18.29 13.026 0.071 6 h 93.26±11.35 94.41±14.33 105.35±16.45 10.856 0.092 8 h 85.33±9.46 97.25±10.54 103.57±11.36 8.451 0.136 10 h 76.25±7.26 71.57±8.33 83.25±9.57 11.733 0.258 12 h 70.25±5.33 67.36±4.22 65.22±3.51 12.528 0.084
3 讨论
阿托伐他汀吸收迅速,2~3 h内即可达到血浆峰浓度,因为其相对较长的半衰期,阿托伐他汀的降脂作用不受一天中给药时间的影响[9]。阿托伐他汀可被细胞色素P450(CYP)3A4和P-450 3A5代谢为邻羟基阿托伐他汀和对羟基阿托伐他汀,这两种活性代谢物延长了阿托伐他汀对3-羟基-3-甲基戊二酰-CoA(HMG-CoA)还原酶的作用,导致酶抑制的半衰期为20~30 h;阿托伐他汀的临床剂量范围为10~80 mg/d,以酸形式给药;阿托伐他汀酸具有高溶解性和渗透性,口服后药物即可完全被人体吸收[9]。小檗碱是从中药小檗碱中提取的一种异喹啉类生物碱,具有多种药理作用[10]。小檗碱通过肠道微生物群的硝基还原酶将自身转化为二氢小檗碱,其在溶液中不稳定,通过氧化在肠组织中恢复为小檗碱。CYP酶是重要的氧化酶,可帮助代谢大部分的临床治疗常用药物[11]。药物代谢分为Ⅰ期代谢和Ⅱ期代谢,CYP酶主要参与药物的I期代谢,其中CYP3A4是关键的同工酶之一,广泛分布于小肠和肝脏,参与临床60%以上药物的代谢[12]。药物对CYP3A4的诱导或抑制可能会影响与它们共同给药的药物的药代动力学,从而改变它们的疗效或毒性。众所周知,小檗碱和他汀类药物通过CYP3A4代谢,竞争结合CYP3A4的活性位点[13]。本研究通过改变在大鼠体内灌胃盐酸小檗碱时间探讨对低剂量阿托伐他汀的药动学影响,结果表明大鼠在单次及多次灌胃小檗碱后,阿托伐他汀体内药代动力学参数的改变无统计学意义(P>0.05)。另外,本研究在给药小檗碱盐酸后,0~12 h内检测了阿托伐他汀在大鼠体内的浓度变化,结果显示在30 min内阿托伐他汀的体内浓度达到峰值,2 h后开始下降,在第12 h维持在60~70 ng/mL。 与A组大鼠相比,B组大鼠和C组大鼠多了盐酸小檗碱的灌胃,但试验结果为盐酸小檗碱对低剂量的阿托伐他汀血药浓度没有影响(P>0.05)。
本研究结果提示临床在联合使用盐酸小檗碱和阿托伐他汀时,正常用量下小檗碱可能不会对阿托伐他汀的药动学造成影响,但由于本研究是在大鼠体内进行的,与人体内的药动学变化是否一致,仍需进一步地研究。
