基于囊性纤维化疾病分子特征及其作用机制的生物信息学分析
2022-10-18王小燕张秋月胡雨洁莫之婧
王小燕,张秋月,胡雨洁,莫之婧
(1.桂林医学院生物技术学院实验教学中心,广西 桂林 541199;2.桂林医学院生物技术学院生物化学与分子生物学教研室,广西 桂林 541199)
囊性纤维化(cystic fibrosis,CF)是白色人种最常见的常染色体隐性遗传疾病之一,主要特征为胰腺功能不全和肺功能渐进性恶化,涉及肺、胰腺、汗腺、消化和生殖系统及其他组织,CF主要由编码囊性纤维化跨膜传导调节因子(cystic fibrosis transmembrane conductance regulator,CFTR)的基因突变引起[1]。欧洲约存在48 000例CF患者,美国约有30 000例CF患者,英国无症状CFTR基因突变的携带者比例约为25∶1,美国约为29∶1[2]。
目前,已有研究[3-6]通过候选基因分析、全基因组关联研究或直接基因测序来鉴定修饰基因,揭示CFTR基因突变与调节液体、电解质运输、细胞内运输和炎症相关基因表达之间的联系,同时CFTR在转录和功能框架中发挥作用。关于CF的分子特征及其作用机制的研究尚未见相关报道,限制了预测单个患者临床病程和治疗反应的能力。目前CF患者的治疗均基于控制疾病临床症状和并发症,而非治疗疾病。因此,阐明CF分子特征及其作用机制并识别可靠的生物标志物对CF的防治至关重要。
微阵列技术和生物信息学分析已被广泛用于筛选疾病相关差异表达基因(differentially expressed genes,DEGs)和功能通路。然而,单一的微阵列分析数据存在假阳性率,难以获得可靠的结果。因此本研究通过对基因表达汇编(Gene Expression Omnibus,GEO)数据库中CF基因芯片联合分析,以期筛选出高可信度DEGs并分析其发挥关键作用的hub基因和生物通路,为CF疾病预测和治疗提供重要参考。
1 材料与方法
1.1 CF基因芯片数据来源使用GEO数据库(http://www.ncbi.nlm.nih.gov/geo/),以“cystic fibrosis”为 检 索 词 检 索。筛 选 条 件:①mRNA表达谱数据集;②以CF为样本;③以正常样本为对照。选择基于GPL570平台检测的GSE71799、GSE24206、GSE98925和GSE69764高通量芯片数据集,采用GEOquery包(2.54.1版本)下载[7]。由GSE71799、GSE24206和GSE98925数据集中获取186个CF样本和37个正常对照样本,由GSE69764数据集中获取3个DNA甲基化抑制剂处理CF样本和3个未经处理的CF样本。将样本数据分为CF组和对照组,根据平台注释信息,将探针转化为相应的基因符号。
1.2 DEGs提取和分析样本中筛除对应多个分子的探针,且仅保留信号值最大的探针,筛除后的数据使用R软件sva包ComBat函数去除批间差,即不同的数据集,使用R软件主成分分析(principal component analysis,PCA)和降维分析(uniform manifold approximation and projection,UMAP)查看样本分组间聚类情况。筛除后的分子总数为21 655个。使用R软件limma包[8]进行2组DEGs分析,筛选标准:P<0.05且|log2(FC)|>1,以log2(FC)>1为CF组 中 显 著 高 表 达 基 因,log2(FC)<-1为对照组中显著高表达基因。采用R软件进行结果可视化,采用R软件ComplexHeatmap包[9]选择聚类方法为欧式距离,绘制2组各前20个DEGs基因表达情况热图。
1.3 功能富集分析使用R软件ClusteProfiler包[10]对 以2组|logFC|>1.5和P<0.05为 阈 值 的DEGs进行基因本体(Gene Ontology,GO)功能注释分析和京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)通路分析,获得DEGs的分子功能(molecular function,MF)、生物学过程(biological process,BP)和细胞成分(cellular component,CC)等注释分析和通路富集分析。使用R软件ClusteProfiler包[10]以分子签名数据库(Molecular Signatures Database,MSigDB)下载C2.cp.v7.0.symbols.gmt作为基因集 富 集 分 析(Gene Set Enrichment Analysis,GSEA)目标集合,获得DEGs显著富集的基因集。计算错误发现率(false discovery rate,FDR)。每个分析将基因集排列重复1 000次计算富集得分(enrichment score,ES)。
1.4DEGs蛋白-蛋白互作(protein-protein interaction,PPI)网络构建和hub基因的筛选使用STRING数 据 库(http://string-db.org)[11]构 建DEGs PPI网络。交互得分阈值0.4作为临界标准。采用Cytoscape软件[12]对PPI网络进行可视化,使用MCODE插件[13]筛选PPI网络中hub基因。筛选标准:MCODE评分>5,度分界=2,节点分界=0.2,最大深度=100,k评分=2。
1.5 统计学分析采用R软件(3.6.3版本)进行统计学分析。2组样本中mRNA表达水平呈正态分布。采用limma包进行2组DEGs分析,以P<0.05为差异有统计学意义。采用ClusteProfiler包对以|logFC|>1.5和P<0.05为 阈 值 的DEGs进 行GO-KEGG通路分析,以P<0.05为差异有统计学意义。采用ClusteProfiler包对DEGs进行GSEA,以P<0.05和FDR<0.25为差异有统计学意义。
2 结 果
2.1 2组样本中DEGs鉴定PCA和UMAP结 果均显示2组样本明显分开并聚为2类,2组间差异明显,后续差异分析有意义的结果可能较多。2组基因表达谱中mRNA表达差异分析结果显示:CF组样本中高表达DEGs 105个,对照组样本中高表达DEGs 324个。DNA甲基化抑制剂未处理CF样本中CXCL2、CXCL3和BEX2为高表达DEGs,其中CXCL2和CXCL3也为对照组样本中高表达DEGs。见图1。
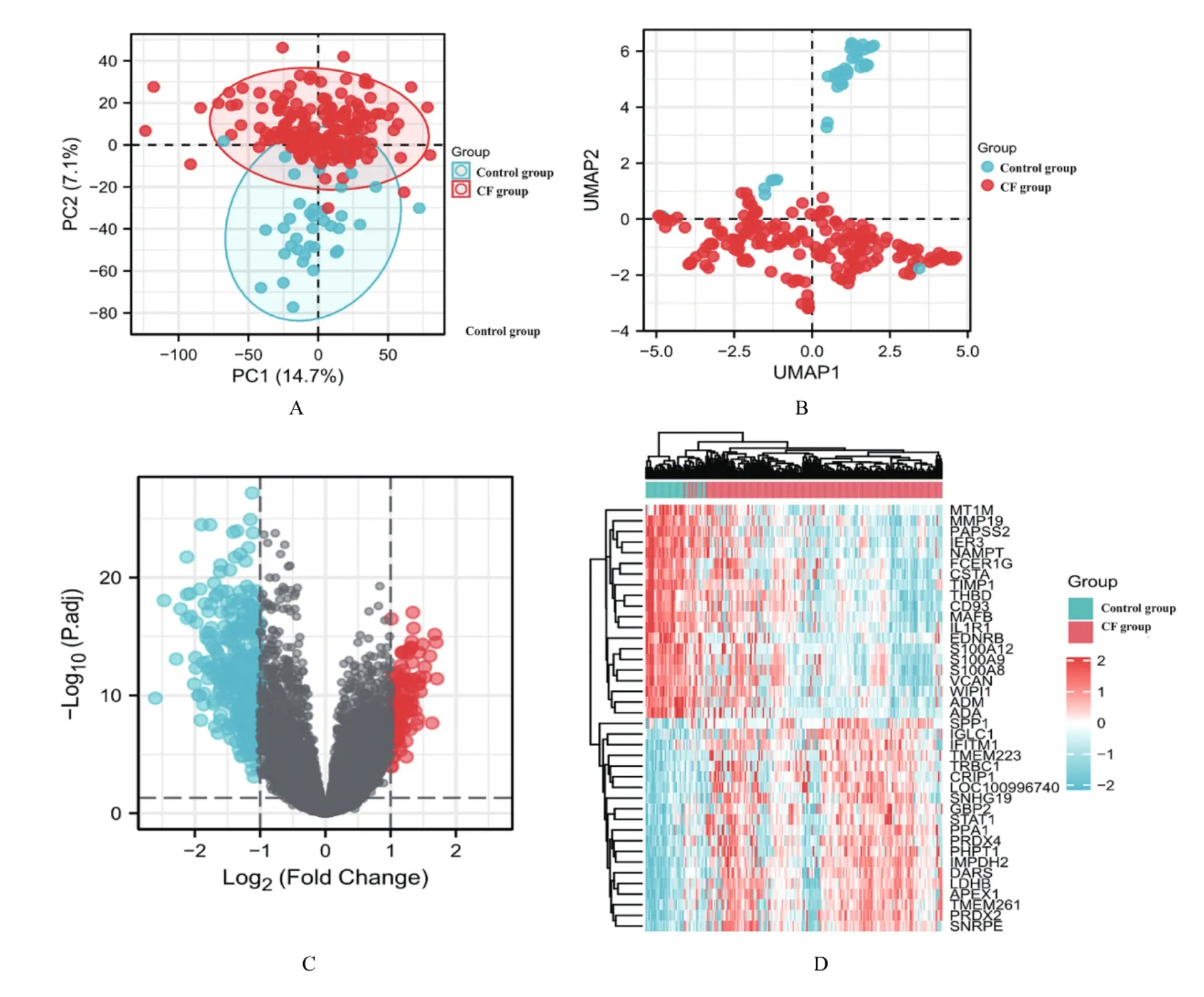
图1 2组样本中DEGs表达情况Fig.1 Expressions of DEGs in samples in two groups
2.2 DEGs富集分析GO-KEGG功能富集分析结果显示:与BP关联显著富集通路共724条,其中前3条分别为中性粒细胞活化(GO:0042119)、中性粒细胞介导的免疫(GO:0002446)和嗜中性粒细胞脱颗粒(GO:0043312);与CC关联显著富集通路共53条,其中前3条分别为胞质囊腔(GO:0060205)、囊腔(GO:0031983)和分泌颗粒内腔(GO:0034774);与MF关联显著富集通路共36条,其中前3条分别为抗氧化活性(GO:0016209)、趋化因子的活动(GO:0008009)和趋化因子受体结合(GO:0042379)。KEGG生物通路显著富集通路共36条,其中前3条分别为类风湿性关节炎(hsa05323)、白细胞介素17(intenleukin-17,IL-17)信号通路(hsa04657)和矿物质的吸收(hsa04978)。见图2A。
GSEA分析结果显示:DEGs在信号通路翻译相关基因集显著富集,其基因主要有PPA1、RPS4Y1和MRPS21等;在核糖体RNA(ribosomal RNA,rRNA)处理相关基因集显著富集,其基因主要有NHP2、RPS4Y1和RPS27L等;在线粒体翻译相关基因集显著富集,其基因主要有MRPS21、MRPL3和MRPS33等;在真核生物翻译延伸相关基因集显著富集,其基因主要有RPS4Y1、RPS27L和RPS5等;在真核生物翻译启动相关基因集显著富集,其基因主要有RPS4Y1、EIF3F和RPS27L等。见图2B和表1。
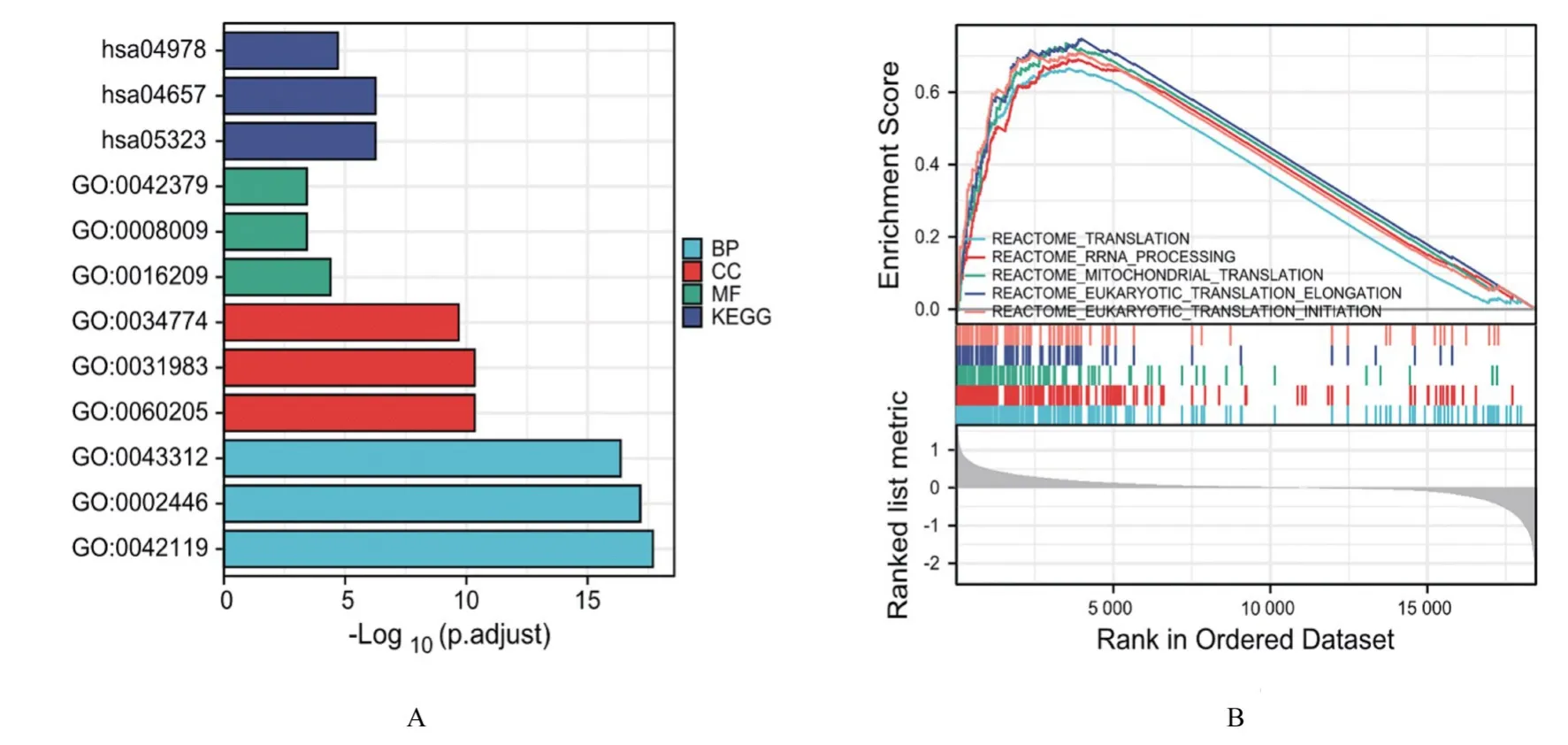
图2 DEGs的GO-KEGG和GSEA富 集分 析Fig.2 GO-KEGG and GSEA enrichment analysis of DEGs
2.3 PPI网络构建和hub基因筛选DEGs相互关联形成复杂的PPI网络,其中最重要的模块包括35个hub基因,分别为基质金属蛋白酶9(matrix metalloproteinase 9,MMP9)、基质金属蛋白酶抑制 剂1(tissue inhibitor of metalloproteinase 1,TIMP1)、血小板反应蛋白1(thrombospondin 1,THBS1)、丝氨酸蛋白酶抑制剂家族A成员1(serpin family A member 1,SERPINA1)、前血小板碱性蛋白(pro-platelet basic protein,PPBP)、C-X-C基 序 趋 化 因 子 配 体2(C-X-C motif chemokine ligand 2,CXCL2)、C-X-C基序趋化因子 配 体3(C-X-C motif chemokine ligand 3,CXCL3)、组织蛋白酶H(cathepsin H,CTSH)、溶菌酶(lysozyme,LYZ)和细胞质FMR1相互作用蛋白1(cytoplasmic FMR1 interacting protein 1,CYFIP1)等。见图3。
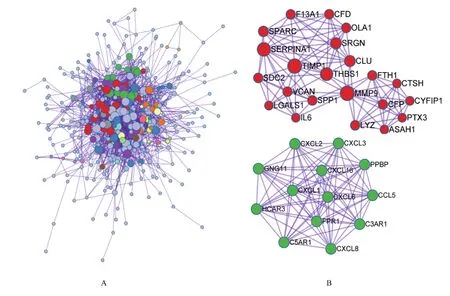
图3 DEGs的PPI网 络 和hub基因Fig.3 PPI network and hub genes of DEGs
3 讨 论
本研究从4个高通量芯片数据集中收集并分析CF样本和对照样本,筛选出429个DEGs,35个hub基因和多条生物通路。DEGs主要富集于与中性粒细胞相关的BP,而中性粒细胞通常被认为是抵御感染的第一道防线,其为人类血液中最丰富的循环白细胞,占白细胞总数约60%,但在CF患者肺毛细血管中存在的中性粒细胞数量增加[14-15]。中性粒细胞是免疫系统武器库中关键的炎性细胞,与细胞因子、趋化因子、白细胞介素受体、集落刺激因子和细胞内信号分子产生相关的约90个基因失调诱导CF患者炎症的发生[16]。有研究[17]显示:功能失调的CFTR由于吞噬溶酶体中次氯酸产生缺陷而损害中性粒细胞杀死病原体的能力。CF患者使用Ivacaftor治疗会导致中性粒细胞迁移和激活减少并促进细菌清除[18]。在与广泛的病原体、促炎症细胞因子或其他炎症信号相互作用后,中性粒细胞被激活并向信息部位移动,通过吞噬作用动员清除入侵的生物体,释放中性粒细胞胞外陷阱(neutrophil extracellular traps,NETs)、细胞因子和趋化因子[19-21]。提示DEGs可能通过调节中性粒细胞细胞免疫影响CF发生发展。
本研究结果显示:DEGs于IL-17信号通路中显著富集。研究[22-23]显示:IL-17协调细胞和有机体的代谢,一方面,IL-17是宿主防御细胞因子,在对真菌和其他细胞外病原体的免疫中发挥重要作用;另一方面,IL-17能驱动多种自身免疫性病理中的炎症。在介导感染免疫过程中,IL-17仅瞬时表达,但在一些慢性炎症和自身免疫性疾病中,IL-17持续长期表达,其影响代谢综合征的能力可能会产生严重后果[23]。细胞水平上,IL-17介导的代谢调节促进细胞增殖,通过上皮细胞、角质形成细胞和干细胞,协调组织修复和伤口愈合[24-26]。某些细胞代谢物(如他康酸盐)会影响IL-17信号转导[27]。因此,推测CF发病机制与IL-17信号通路密切相关,该信号通路上的基因可能是CF的关键基因。
GSEA分析结果显示:DEGs主要在翻译的启动、延伸和信号通路翻译等基因集中富集。研究[28]显示:NHP2的功能缺陷影响rRNA的生物发生,并导致肺纤维化。RPS4Y1与慢性阻塞性肺疾病相关[29]。PPA1作为一种新的PPARγ靶基因,通过维持脂肪细胞线粒体功能调节全身胰岛素敏感性[30]。MRPS21与胰岛自身免疫进展为Ⅰ型糖尿病相关[31]。本研究结果显示:与肺和胰腺功能相关基因均为CF关键通路的基因,而CF以胰腺功能不全和肺功能渐进性恶化为特征,提示相关基因可能在CF中发挥着重要的作用,可能是CF潜在生物标志物。本研究结果显示:RPS27L、MRPL3、MRPS33、RPS5和EIF3F等 基 因 与CF有关,为探讨CF的分子机制提供了新的思路。但DEGs与CF的发生发展之间的联系,仍需要进一步实验验证。
本研究筛选出35个hub基因,可能为CF发生发展过程中的关键基因。研究[32]显示:与非CF单核细胞比较,CF患者单核细胞中MMP9表达水平升高。MMP9可以提高胶原蛋白脯氨酸-甘氨酸-脯 氨 酸(proline glycine proline,PGP)的 水 平,CF患儿肺部PGP水平升高可能加剧炎症反应[33]。而其他34个hub基因在CF中的作用尚未阐明,本研究结果为CF分子机制相关研究提供了方向。未使用DNA甲基化抑制剂处理的CF样本中高表达CXCL2和CXCL3,其在正常对照样本中亦高表达,且均包含于35个hub基因中。研究[34]显示:遗传和表观遗传学的改变在纤维化疾病中发挥作用,如DNA甲基化可以改善小鼠肺纤维化,提示DNA甲基化可能是CF治疗的重要方向,CXCL2和CXCL3可能与CF发生发展有密切关联,有望成为CF的生物标志物。
综上所述,本研究揭示了CF相关DEGs、hub基因和生物通路,为探讨CF分子机制提供了新的研究方向和思路,为阐明hub基因的生物学功能和CF的标志物治疗提供了理论依据。
