简易合成天然修饰核苷2′-脱氧古鸟苷
2022-10-18吴明智
匡 爽,彭 静,田 佳,吴明智,明 新
(成都医学院药学院,四川 成都 610500)
0 引言
天然修饰DNA不像天然修饰RNA那样化学种类繁多、普遍存在.到目前为止,已发现的天然修饰DNA约有十几种[1-2].在细菌中发现的硫代磷酸酯修饰的DNA骨架在不同的生物体中发挥着不同的功能[3-6].这些修饰的DNA似乎有特殊的标记,以避免被自身的酶系统降解,并可以在调节基因表达(如热稳定性和转录调节等)方面发挥关键作用[7].L.M. Iyer等[8]通过计算机预测,提出在细菌和噬菌体中有十几种新型DNA修饰系统,这表明DNA修饰的潜在多样性和复杂性还有待检测.7-脱氮嘌呤(7-deazapurine)作为微生物的代谢物天然存在于自然界中[9],由于其广泛的生物活性而受到人们的关注[10].如7-脱氮-2′-脱氧鸟苷和7-脱氮-2′-脱氧腺苷是有效的端粒酶抑制剂[11],它们能被DNA聚合酶识别而链入DNA中,并被用于DNA测序[12-14].由于7-脱氮嘌呤与天然嘌呤结构相似,所以它们可与嘧啶形成稳定的碱基对[15].7-氰基-7-脱氮-2′-脱氧鸟苷(dPreQ0)和7-氨基-7-脱氮-2′-脱氧鸟苷(dADG)首次在肠道沙门氏菌血清蒙得维的亚的DNA酶水解产物中被发现[16],最近又分别在噬菌体和细菌DNA中被发现[17].PreQ0是由GTP经合成酶(FolE、QueD、QueE和QueC)合成后再进行转糖基化得到的,而dG+是由PreQ0在酶作用下转化而来的.本文用化学方法制备天然修饰的7-甲脒基-7-脱氮-2′-脱氧鸟苷(2′-脱氧古鸟苷,dG+),以进一步研究这些修饰核苷在生物合成过程中以及在DNA和RNA之间的作用.优化了dG+关键前体dPreQ0的合成,报道了dG+的化学合成,并通过核磁共振和HR-MS证实了其结构(见图1).天然修饰DNA的合成将为生物技术的发展和新型抗菌剂的开发提供支持.
1 实验部分
1.1 主要仪器与试剂
实验所用仪器有:核磁共振波谱仪,Avance 400型(DMSO为溶剂,TMS为内标),瑞士布鲁克公司;Agilent 6230B液质联用仪,安捷伦科技(中国)有限公司.实验所用试剂均为市售分析纯.
1.2 合成方法
1.2.1 关键中间体dPreQ0(3)的合成 4-氯-5-碘-2-特戊酰氨基-7H-吡咯并[2,3-d]嘧啶(3)的合成:将5.00 g(29.66 mmol)2-氨基-4-氯吡咯并[2,3-d]嘧啶(1)溶解在100 mL吡啶中,在冰浴冷却下缓慢滴加10.70 g(88.98 mmol)特戊酰氯,并使混合物在室温下反应12 h.待反应结束后,在冰浴冷却下向反应体系中添加250 mL饱和NaHCO3水溶液,并搅拌30 min.然后将混合物用二氯甲烷(60 mL×3)萃取,合并有机部分,用盐水洗涤并用无水Na2SO4干燥.真空浓缩有机部分,得到化合物2,为黄色粉末.再将5.96 g(26.67 mmol)N-碘琥珀酰亚胺(NIS)加入搅拌的化合物2(6.11 g,25.71 mmol)的二氯甲烷(80 mL)溶液中,并在室温下反应3 h.待反应完成后,将混合物用1 mol·L-1的H2SO3洗涤至无色.将有机部分浓缩至干,残余物用乙醇重结晶即得黄色粉末状的化合物3(7.97 g,产率70.98%,2步).Rf=0.80(V(CH2Cl2)∶V(MeOH)=20∶1);1H NMR(400 MHz,DMSO-d6):12.73(s,1H,NH),10.14(s,1H,CONH),7.79(s,1H,6H),1.24(s,9H,3CH3).该数据与文献[18]相同.HR-MS:378.982 3([M+H]+,C11H13ClN4O+;cald 378.981 7).
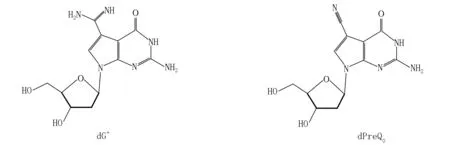
图1 dG+和dPreQ0的化学结构

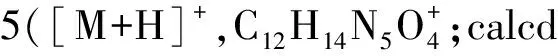
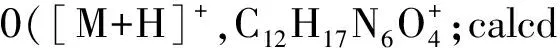

图2 dG+1H-NMR谱图

图3 dG+13C-NMR谱图

图4 dG+ MS谱图
2 结果与讨论
dPreQ0是2′-脱氧古鸟苷的关键中间体,dPreQ0的合成最早由文献[20]报道,以2-氨基-4-氯吡咯并[2,3-d]嘧啶为底物(1),合成路线如图5所示[21].每个化合物都用柱色谱法纯化,耗费了大量的时间和溶剂.因此,本文对dPreQ0及其中间体的合成进行了优化.
根据文献[20]报道,化合物3由2-氨基-4-氯-吡咯并[2,3-d]嘧啶先经特戊酰氯酰化,再经N-碘代琥珀酰亚胺碘化得到,2步反应均采用硅胶柱分离纯化.由于碘代物溶解性差,用色谱法纯化会消耗大量溶剂,因此本文采用工业上最常用的萃取和重结晶方法来分离纯化.在酰化反应完成后用二氯甲烷萃取,有机相经蒸干后直接用于碘代反应,避免了过柱纯化.在碘代反应完成后用稀亚硫酸钠水溶液淬灭剩余的NIS,用甲醇重结晶,2步的产率为60.8%.若改用乙醇重结晶,则产率为70.9%,文献[21]的产率为71.4%(化合物2的产率为84.0%,化合物3的产率为85.0%).本文通过萃取和乙醇重结晶得到目标产物,该纯化方法减少了有机溶剂的使用和时间的消耗.
7-碘-2′-脱氧-7-脱氮鸟苷(6)是dPreQ0的前体.化合物6的合成路线(见图6)主要有2条:(i)将化合物4与甲醇钠进行亲核取代反应,然后再与2 mol·L-1的NaOH进行反应[22];(ii)用Cs2CO3、三乙二胺(TED)和Et3N将化合物4的4位氯基变成羰基,然后在强碱条件下除去保护基[23].与路线(i)相比,路线(ii)所需试剂昂贵且合成时间更长,因此,本文采用合成路线(i)制备化合物6.将4-氯-5-碘-2-特戊酰基氨基-7H-吡咯并[2,3-d]嘧啶(3)与1-氯-3,5-二-(对-甲苯氧基)-2-脱氧核糖进行糖基化,得到β-D-核苷(4),将化合物4在0.03 mol·L-1的甲醇钠中回流制备化合物5.对于这些化合物,在文献[22]中每步都通过硅胶柱色谱法纯化.本文实验发现将糖苷化反应混合物pH值调节为7,并用二氯甲烷萃取可以得到粗产物4.它可以直接用于合成化合物5,并且化合物5可以用乙酸乙酯从反应液中萃取出来,直接用于化合物6的制备.由于化合物6的极性较高,用硅胶柱分离会消耗大量溶剂,所以本文选择用树脂AB-8代替硅胶色谱,以纯水和乙醇为流动相.

注:试剂和条件为(a)PivCl,Py,0 ℃,12 h;(b)NIS,DCM,r.t.,3 h;(c)NaH,MeCN,r.t.,2 h;(d)NaOMe,MeOH,reflex,3 h;(e)2 mol·L-1 NaOH,reflux,2 h;(f)CuCN,Py,120 ℃,7 h.

图6 7-碘-2′-脱氧-7-脱氮鸟苷的合成路线
在合成化合物dPreQ0时,考虑到氰化亚铜的毒性及其对环境的污染,决定减少它的用量,设置不同当量进行反应.实验发现:将氰化亚铜的当量从7降低到5可以基本保持产率不变,合理减少了氰化亚铜的用量(见图7).
为了将氰基转变为甲脒基制备2′-脱氧古鸟苷,首先想到Pinner反应.文献[24]用该反应制备古鸟苷,得到古鸟苷和三乙胺醋酸(2∶1)的混合物,产率为30%.经典的Pinner反应是在HCl气体催化下,腈基首先醇解得到亚胺酸酯中间体,然后氨解得到产物.但通HCl气体,操作方法烦琐,并对设备会造成很大的腐蚀.因此,笔者尝试用甲醇钠作碱来催化制备亚胺酸酯中间体.实验发现用碱催化反应条件虽然温和,但是原料在室温下4 d也不能反应完全.因此,对通HCl气体催化的方法进行了优化,其过程如下:在冰浴冷却下,将乙酰氯滴加至甲醇中产生HCl催化氰基转化为亚胺酸甲酯,然后该中间体在3.5 mol·L-1氨甲醇中反应过夜,在反应结束后,挥去氨气,2′-脱氧古鸟苷直接从甲醇中析出得到目标化合物2′-脱氧古鸟苷,产率为92.59%.合成路线如图8所示.
3 结论
本文用简单有效的方法成功合成了天然核苷dG+,并对关键中间体dPreQ0的合成工艺进行了优化.以2-氨基-4-氯吡咯并[2,3-d]嘧啶为起始物,经7个步骤合成dG+的总产率为33%,并以更环保的方式获得了中间体.dPreQ0和dG+的合成为天然修饰DNA的合成提供了一条有价值的途径.
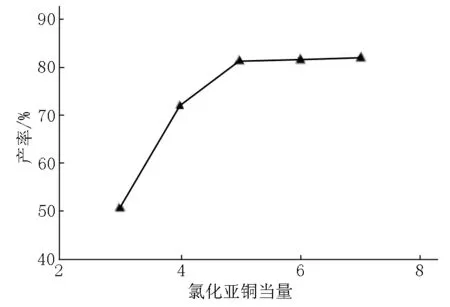
图7 氰化亚铜当量对dPreQ0产率的影响
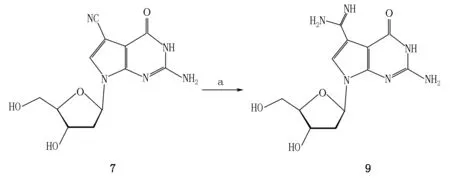
注:试剂和条件为(a)AcCl,MeOH,r.t.,6 h;3.5 mol·L-1 NH3/MeOH,r.t.,overnight.
