遗传性心脏病死后基因检测技术的研究进展及法医学应用
2022-10-14董怡铭杨琛腾张国忠丛斌
董怡铭,杨琛腾,张国忠,丛斌
河北医科大学法医学院 河北省法医学重点实验室 河北省法医分子鉴定协同创新中心,河北 石家庄050017
据推算,目前我国心血管病患病人数达到3.30亿,心血管病死亡占城乡居民总死亡原因的首位,在农村为46.66%,城市为43.81%[1]。近年来我国心脏性猝死(sudden cardiac death,SCD)案例不断增加,SCD 的发生率因研究对象的年龄而异,其病因也与年龄有关[2]。冠心病和急性心肌梗死是老年人SCD最常见的原因,在青壮年人群(<35 岁)中,遗传性心脏病及感染性心肌炎是SCD 的主要原因。遗传性心脏病主要包括离子通道病与遗传性结构性心脏病两大类,具有高度的SCD 风险。遗传性结构性心脏病主要为遗传性心肌病,可导致心律失常、心力衰竭和SCD。部分遗传性结构性心脏病可通过尸体检验确定死因。相比之下,单纯致心律失常的离子通道病更难确定死因,因为这些疾病不会导致心脏结构的改变,尸体检验时组织病理学检验和毒(药)物检验常无阳性发现,造成死亡原因难以确定,通常被归类为“死因不明”,这种情况在年轻人SCD 案例中多达30%[3],因此,该类死者是否存在潜在的遗传病值得探讨。
死后基因检测技术是应用分子生物学技术对死者进行基因学检测,当常规尸体检验不能明确死因时,可在尸体检验时采集血细胞或组织细胞,提取DNA 进行全基因或目的基因检测,这种在法医病理学实践中进行死因鉴定的技术手段称为死后基因检测技术[4]。死后基因检测技术对遗传性心脏病猝死病因的诊断具有重要作用,可以确定SCD 病例超过30%的死因[3]。通过对死者进行有针对性的基因检测,可以提高猝死原因的诊断准确性。本文就近年来国内外关于离子通道病和遗传性心肌病的类型、发病机制及分子遗传学相关研究进行总结,并探讨死后基因检测技术在法医学检案中的应用,以期为SCD 的法医病理学诊断及鉴定提供依据。
1 离子通道病与SCD
离子通道病是指无器质性心脏病却以心电紊乱为主要特征的一类疾病,多数为常染色体显性或隐性遗传,呈家族多发倾向,也可散发,临床上可出现晕厥、抽搐甚至导致SCD,可在任何年龄发病[5],主要是由参与产生心脏动作电位的离子通道分子缺陷引起[6]。离子通道病相关的易感基因见表1~2。离子通道病主要包括长QT 间期综合征(long QT syndrome,LQTS)、儿茶酚胺敏感性多形性室性心动过速(catecholaminergic polymorphic ventricular tachycardia,CPVT)、Brugada 综合征(Brugada syndrome,BrS)、特发性心室颤动(idiopathic ventricular fibrillation,IVF)、短QT 综合征(short QT syndrome,SQTS)、进行性心脏传导障碍(progressive cardiac conduction defect,PCCD)等心脏传导性疾病。
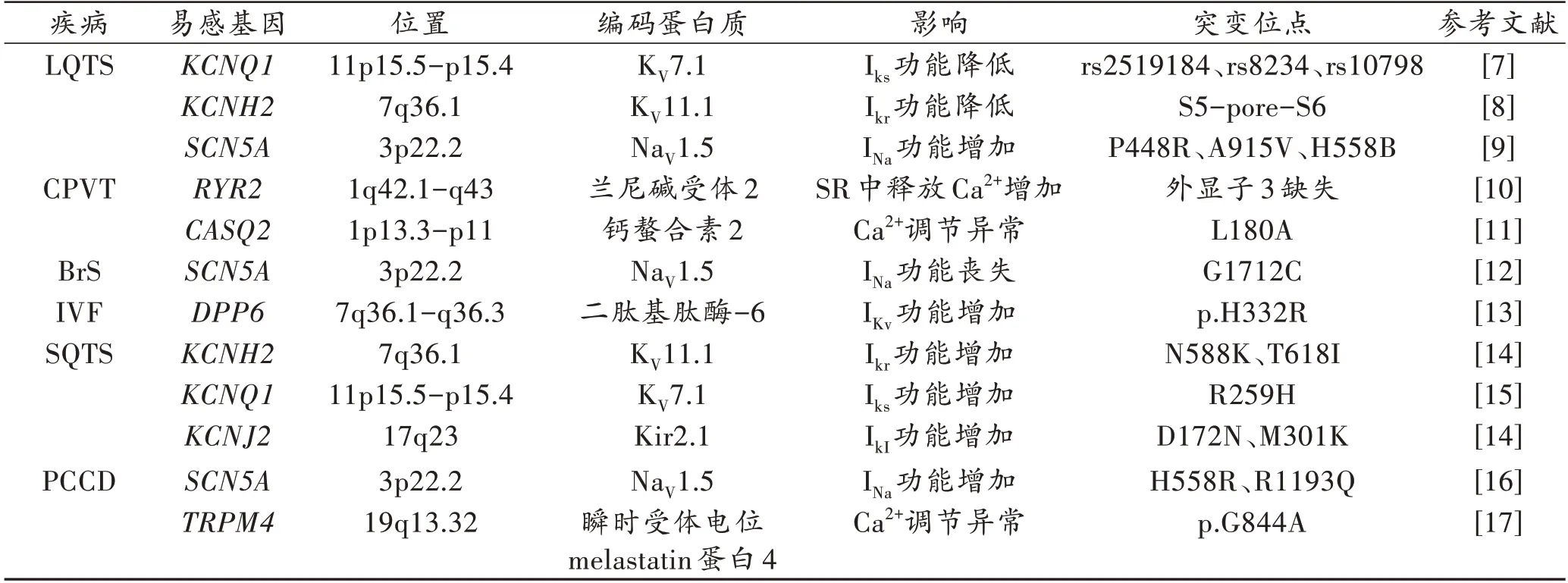
表1 离子通道病主要易感基因Tab.1 Main susceptibility genes of channelopathies
1.1 LQTS
LQTS 指心电图上QT 间期延长、T 波异常,易产生室性心律失常,导致反复发作性晕厥甚至猝死的一组综合征。LQTS 具有明显家族聚集性,约70%的病例由基因缺陷所致[30]。SCD 可能是LQTS 的第一个表现,而且LQTS 是最常见的遗传性心律失常综合征。回顾性分析香港地区59 例LQTS 患者发现,平均发病年龄为8.2 岁,男性占56%,8.5%合并先天性心脏病,1 年、5 年和10 年的心脏事件发生率分别为93.0%、80.7% 和72.6%[31]。在中国西南地区,33.7%的不明原因猝死的患者携带LQTS 相关突变基因[31]。

表2 离子通道病罕见易感基因Tab.2 Rare susceptibility genes of channelopathies
近25 年来,已发现在多达75%的LQTS 病例中,17 个编码心脏离子通道、离子通道辅助亚基或调节离子通道功能的蛋白质的基因发生致病突变[32]。美国国立卫生研究院成立的临床基因组资源工作组报告 的17 个LQTS致病基因中,9个基因(AKAP9、ANK2、CAV3、KCNE1、KCNE2、KCNJ2、KCNJ5、SCN4B、SNTA1)被归类为证据有限或有争议的LQTS致病基因,只有3 个基因(KCNQ1、KCNH2、SCN5A)被确定为典型的LQTS 致病基因,另外发现4 个基因(CALM1、CALM2、CALM3、TRDN)有明确或强有力的证据支持其在引起LQTS 中的作用,但具有特定的非典型特征[8-9,18]。在所有LQTS 亚型中,LQT1~3 最常见,分别为KCNQ1、KCNH2和SCN5A基因缺陷所致[7]。KCNQ1基因3′非编码区的SNP 可有效改变LQT1 的疾病严重程度[33]。国外1 项研究[7]在所有LQT1 患者KCNQ1基因3′非编码区中发现6 个SNP,其中rs2519184、rs8234 和rs10798 以等位基因特异性方式与QT 间期和症状发生相关。KCNE1和KCNE22 个基因,除了被报道为先天性LQTS 的致病基因外,还有文献[18]报道其在药物或电解质引起的LQTS(即获得性LQTS)病因中发挥作用。三体蛋白是另一种参与心肌细胞钙依赖过程的蛋白,包括钙释放和兴奋收缩偶联的调节。ALTMANN 等[19]通过对33 名LQTS 患者进行全外显子组测序,并将TRDN确定为隐性遗传LQTS 的新的潜在遗传基础,所有确诊病例的不典型特征包括常染色体隐性遗传,儿童早期表现为多次发作的劳力诱发的晕厥或心搏骤停,常见的ECG 特征是胸前导联V1~V4 广泛T 波倒置,具有一致或短暂的QT 延长,以及骨骼肌无力的可能性。生前符合这些表型特征的死者应接受心脏CALM1~3和TRDN基因检测。在成人病例中,这些基因在导致LQTS 更典型表现中的作用仍有待确定。其他与LQTS 密切相关的基因有NOS1AP、CACNA1C[20-21]。
1.2 CPVT
CPVT 是一种由肾上腺素诱导的室性心律失常伴晕厥和猝死为特征的家族性疾病,平均发病年龄为7~9 岁,人群中发病率约为1/10 000,心搏骤停可能为CPVT 的首发症状,是儿童和青少年猝死的重要原因之一[34]。患者有正常的心脏结构和收缩功能,静息状态下心电图正常,但在交感肾上腺系统兴奋(运动或情绪激动)时诱发双向或多形性室性心动过速,可恶化为心室纤颤,临床表现主要为晕厥和心搏骤停。据报道[10],5种基因(RYR2、CASQ2、CALM1、TRDN和TECRL)的突变与CPVT 表型相关。RYR2基因点突变可导致CPVT1,表现为常染色体显性遗传[35]。在确诊为CPVT 的95%患者中发现,RYR2基因的功能获得性突变[36]。RYR2编码一个相对分子质量565 000 的同源四聚体蛋白质,构成心肌肌质网膜上的Ca2+通道[37]。静息状态下,肌浆网正常释放Ca2+,但在运动或情绪激动时,肌浆网内Ca2+浓度突然增加,当RYR2基因突变时,该通道对Ca2+的敏感性增加或阈值降低,细胞质内较低浓度的Ca2+即可激活Ca2+通道,诱发Ca2+的大量释放,细胞内Ca2+浓度增加,从而导致延迟后除极与触发活动[37]。虽然关于RYR2突变导致CPVT 的原因有多种假设,但多数人认为CPVT 相关的RYR2突变增加了舒张期间RYR2自发开放和病理性钙释放[11]。国内有研究[38]纳入12 例CPVT 患者,发现平均发病年龄(8.4±3.2)岁,2/3 为男性,RYR2突变占75%,平均随访(0.92±0.80)年后发现1 例患者死亡。CASQ2突变较罕见,点突变导致CPVT2,可解释某些临床少见的常染色体隐性遗传的CPVT[35]。但GRAY 等[39]对一个家族中诊断为显性遗传的严重CPVT的6 名成员进行研究,外显子组测序鉴定了CASQ2(Lys180Arg)中一个新的杂合错义突变,影响一个高度保守的残基,该残基与疾病共分离,在未受影响的家族成员中缺失,并首次发现CASQ2变异可引起常染色体显性CPVT,并提出显性遗传的CPVT 的遗传检测应包括CASQ2杂合变异的筛查。其他与CPVT 相关的基因有TRD、ANK2[22]。
1.3 BrS
BrS 是一种常染色体显性遗传病,与年龄和性别相关,其特征是心前区导联ST 段持续抬高(V1~V3),右束支传导阻滞,易发生室性心律失常和SCD。BrS的患病率在1/5 000~1/2 000,心室颤动发生在(41±15)岁,但心室颤动可能在任何年龄出现,通常在休息或者睡眠期间,男性的患病率是女性的8~10 倍[23]。BrS 在结构正常的心脏病患者中有较高的SCD 发生率,该病引起的猝死占所有SCD 的4%~12%,占无器质性心脏疾病猝死者的20%[23]。第一个被报道与BrS 相关的基因是SCN5A基因,其编码心脏钠通道的α 亚单位,SCN5A基因突变导致钠通道功能丧失[12]。编码心脏钠通道的相关基因有SCN1B(编码钠通道β-1 亚单位)、SCN2B(编码钠通道β-2 亚单位)和SCN3B(编码钠通道β-3 亚单位),所有这些突变都改变了通道的功能(增加或减少Na+电流),与BrS 发生密切相关。VAN DEN BOOGAARD 等[40]通过去除SCN10A上 的有关序列,发现SCN5A的表达明显减少,因此,SCN10A可能通过调节SCN5A的功能从而在心律失常和SCD 中发挥重要作用。另一个负责BrS 的基因是GPD1-L,GPD1-L基因突变导致编码的蛋白第112 位脯氨酸(Pro,P)被亮氨酸(Leu,L)取代会降低心脏Na+电流和在细胞膜附近GPD1-L的表达[41]。GPD1-L可能影响心脏Na+通道向细胞膜表面运输Na+。GPD1-L突变降低了SCN5A表面膜表达,降低内向Na+电流,并导致BrS[23]。其他与BrS 密切相关的基因有SCN1B、SCN2B、PKP2、KCNJ8、KCND3、ABCC9、HCN4、CACNA2D1、TRPM4等[23]。
1.4 IVF
IVF 是指患者发生心室颤动甚至SCD 后,经详尽的临床检查未发现明显心脏结构异常或已知相关遗传学异常的一类疾病。目前IVF 主要是基于临床现象的一种排除性诊断。IVF 的临床特征为早期复极,表现为前壁或侧壁导联J 点抬高及ST 段抬高,通常被认为是一种良性改变,常见于青少年,多发于男性,部分突变在女性携带者上无明显临床症状及心律失常事件发生[42]。原因可能是女性的XX 染色体中的一条失活可导致突变基因功能部分丧失。根据既往研究[43],二肽基肽酶-6(dipeptidyl peptidase-6,DPP-6)和心脏钠通道NaV1.5(SCN5A)的突变是IVF 中最重要的遗传因素。DPP-6是心肌细胞瞬时外向钾通道的调控单位,由N 末端、跨膜结构域、糖基化结构域、富含半胱氨酸结构域和氨基肽酶结构域组成。DING等[13]采用全外显子组测序结合心律失常相关基因筛选的方法,发现了一个新的DPP-6错义突变(c.1578G>c/p.Q526H),并通过膜片钳实验发现,当DPP-6 过表达时可以导致浦肯野纤维外向钾电流增多,这就解释了DPP-6突变致心律失常的潜在机制。也有文献[44]报道,在复发性心室颤动患者中检测到DPP-6错义突变(p.H332R)。上述两种突变都可以导致电压门控钾通道(Kv4.3)的功能增强并导致心室颤动。据相关文献[24-27]报道,与IVF 的发生密切相关的突变基因还有IRX3、CALM1-3、CACNA1C、KCNJ8等。
1.5 SQTS
SQTS 是临床上不常见的一种遗传性离子通道病,容易诱发心房颤动、室性心动过速和SCD。SQTS属于常染色体显性遗传,主要由钾-钙离子通道功能缺陷导致QT 间期异常缩短以及室性和房性心律失常风险增加引起[45]。34%的患者表现为SCD,15%的患者有SCD 家族史[46]。基因检测对于诊断该疾病很重要,但迄今为止,致病突变的发现率低于25%[46]。目前,已经发现导致SQTS 的6 个离子通道基因发生突变,分别是KCNH2(SQT1)、KCNQ1(SQT2)、KCNJ2(SQT3)、CACNA1C(SQT4)、CACNB2(SQT5)和CACNA2D1(SQT6)[14]。编码快速延迟整流钾通道(human ether-α-go-go related gene,hERG)的KCNH2基因中的功能获得突变N588K 已被证明是SQT1 形式的SQTS 的基础[14]。有研究[15]克隆了R259H 并在人胚肾细胞(human embryonic kidney cells,HEK)-293 中瞬时表达,然后通过全细胞膜片钳技术记录电流,发现R259H-KCNQ1电流密度显著增加,在1 s 的去极化脉冲后,电流密度约是野生型的3 倍,其结果表明,R259H 是负责SQTS2 的KCNQ1通道的功能获得性突变,这是首次在中国人中检测到R259H 突变。KCNJ2基因突变会导致延迟整流钾电流(IKr)功能亢进,该突变导致心房-心室有效不应期和QT 间期缩短,这将增加心室颤动和心房颤动的易感性[45]。
1.6 PCCD
PCCD 是一种遗传性心脏病,其特征是通过房室束-浦肯野系统的冲动传导进行性延迟伴左或右束支传导阻滞,病情严重时可发生完全性房室传导阻滞、晕厥和SCD[47]。PCCD 是年轻人和老年人群中相对常见的疾病,但也有在婴幼儿期和儿童期发病的案例报道[17,48]。在心脏结构正常的情况下,家族性PCCD 通常是由编码心脏离子通道的基因突变引起的,而在结构性心脏病的情况下,PCCD 通常是由编码转录因子、蛋白激酶或结构蛋白的基因发生突变引起的[47]。目前研究[16]发现,结构正常心脏中遗传性PCCD 的发病机制主要与离子通道基因(如SCN5A、SCN1B、SCN10A、TRPM4和KCNJ2)以及编码心脏连接蛋白的基因(如GJA5)中的遗传变异有关。编码心脏钠通道NaV1.5 的SCN5A是已知导致家族性PCCD 的主要离子通道基因[16]。SCN5A突变导致的PCCD 通常可以与其他遗传性心脏病重叠或共存,如3 型LQTS(LQT3)、1 型BrS(BrS1)等[28]。近期研究[17,47]发现,PDYN基因中两个相邻的错义突变[c.581A>T,c.580G>C(p.D194L)]和TRPM4基因外显子17 中的p.G844A 突变可能与家族性PCCD 的发病有关。其他与PCCD 密切相关的基因还有LMNA、GJA5、CACNA1C、HCN4、TBX3、TBX5、PRKAG2等[16,28-29]。
此外,其他一些离子通道病也具有SCD 高风险,如早期复极综合征、特发性右心室流出道室性心动过速等,可能还包括青壮年猝死综合征和婴儿猝死综合征等[49-52]。
2 遗传性心肌病与SCD
心肌病是一种由心肌功能异常引起的异质性疾病,可导致心律失常、心力衰竭和SCD,其可分为原发性心肌病(遗传性、混合性或获得性)和继发性心肌病[53]。根据其表型特征分为肥厚型心肌病(hypertrophic cardiomyopathy,HCM)、扩张型心肌病(dilated cardiomyopathy,DCM)、限制型心肌病(restrictive cardiomyopathy,RCM)、致心律失常性右心室心肌病(arrhythmogenic right ventricular cardiomyopathy,ARVC)和未分类型心肌病[54]。遗传性心肌病的发生和发展均与单个或多个基因突变有关[55]。遗传性心肌病相关的基因见表3~4。

表3 遗传性心肌病主要易感基因Tab.3 Main susceptibility genes of hereditary cardiomyopathy
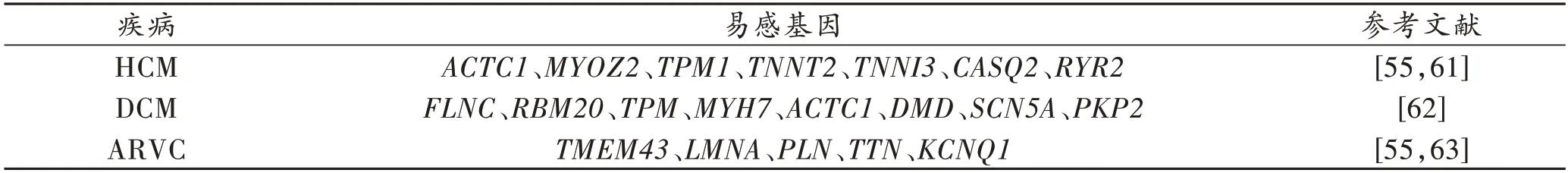
表4 遗传性心肌病罕见易感基因Tab.4 Rare susceptibility genes of hereditary cardiomyopathy
2.1 HCM
HCM 是最常见的常染色体显性遗传的心血管疾病,致病基因携带者的患病率达1/500,甚至1/200[64],是青少年、竞技运动员发生SCD 的首要原因[57]。临床特征以非对称室间隔肥厚、心室腔变小,左心室腔充盈受阻以及舒张期顺应性下降为特征,常伴有左心流出道狭窄。室间隔与左心室厚度比值由正常的0.95增加到1.3 以上,此为诊断HCM 主要指征。HCM 的主要组织病理学改变为肌细胞排列紊乱、细胞外基质改变、微血管功能障碍和心肌纤维化等[65]。HCM 是一种主要由编码心肌肌小节结构蛋白的基因发生突变而导致的单基因遗传病,以常染色体显性遗传为主(约占疾病的2/3),遗传异质性突出[66]。有研究[67]表明,约7%的HCM 患者存在多个致病基因突变或复合突变,通过基因型与SCD 风险关联分析发现,突变基因存在量效关系,即多基因或复合突变的患者较单突变患者发病年龄早、SCD 风险大。HCM 的遗传基础已经部分阐明,MYH7和MYBPC3分别编码β-肌球蛋白重链和肌球蛋白结合蛋白C,是两个最常见的致病基因,占所有导致HCM 致病基因的50%[57]。MYH7基因编码的β-肌球蛋白重链是肌小节的主要收缩蛋白。MYH7突变引起HCM 的表型具有发病早、外显率高、心肌肥厚程度重、猝死率高、临床表型比MYBPC3突变严重的特点[68]。有研究[56]发现,在已经被诊断为HCM 的患者中,高通量测序发现,MYH7-V934A、MYH7-E1387K 和MYH7-M877I为引起HCM的新型MYH7突变。MYBPC3基因编码心肌肌球蛋白结合蛋白C(cardiac mysoin-binding protein C,cMyBP-C),是粗肌丝的重要组成部分,通过不同激酶的磷酸化调节肌动-球蛋白相互作用。MYBPC3突变引起HCM的表型通常具有低外显率、晚发病、良性进程的特点,大多为杂合突变,罕见的纯合突变会导致积累效应,加重HCM 的临床表型和SCD 发生率[69]。其他与HCM发生密切相关的突变基因有ACTC1、MYOZ2、TPM1、TNNT2、TNNI3、CASQ2和RYR2等[55,61]。
2.2 DCM
DCM 是一种原因未明的原发性心肌疾病,特征为左或右心室或双侧心室扩大,并伴有心室收缩功能减退,最终导致心力衰竭。此外,DCM 还会使患者易患心律失常性SCD[70]。DCM 在心力衰竭人群中的患病率为1∶400~1∶250,在普通人群中为1∶2 500[71]。大多数患者在20~60 岁出现症状,但在儿童也可能出现(DCM 占儿童心肌病的60%)[72]。中国心力衰竭注册登记研究纳入2012—2015 年132 家医院的13 687 例心力衰竭患者,16%的患者为DCM[73]。DCM 的病因可分为遗传性和非遗传性,但两组之间存在重叠。由编码细胞骨架、肌节或核膜蛋白的突变基因引起的遗传性DCM 约占所有DCM 病例的35%[72]。大多数这些基因突变作为常染色体显性性状遗传,X 连锁遗传、常染色体隐性遗传和线粒体模式的遗传很少见[74]。TTN截断变异是DCM 最常见的遗传原因,占所有病例的14%~25%[75]。通过对具有TTN突变的DCM 患者的基因型-表型关联的回顾性研究[75]发现,DCM 表型在具有TTN截断变异的受试者中的外显率随着年龄的增长而增加,在40 岁时达到95%,并在70 岁时达到100%。该基因位于2 号染色体(2q31.2)上,编码肌联蛋白,TTN基因的大多数突变都位于A 带区域,该域对于肌联蛋白作为生物分子支架的功能至关重要[76]。已有研究[77]表明,过度酗酒或病毒感染会加剧截断肌联蛋白突变体的有害影响,表明遗传和环境风险因素之间可能存在相互作用。5%的DCM 病例是基于1 号染色体短臂上的LMNA基因突变,LMNA编码核纤层蛋白A/C,属于核纤层蛋白家族,是维持正常核膜形态的主要细胞骨架蛋白[78]。通过比较DCM 患者和健康对照者rs4641位点发现,在DCM组中,rs4641 的TC、TT 基因型频率和T 等位基因频率均高于对照组(P<0.01)。因此得出,LMNArs4641 位点的C 到T 突变可以增加DCM 的风险,表明LMNA是DCM 的易感基因[59]。心肌肌钙蛋白T 型-2(TNNT2)基因位于1q32 区域,编码将肌钙蛋白复合物连接到原肌球蛋白上的收缩蛋白[79]。DCM 患者TNNT2基因片段分析结果发现,TNNT2基因中的错义突变(Leu84Phe)和两个新的SNP(c.192+353C>A,c.192+463G>A)可能与中国人群的DCM 相关[60]。其他与HCM 发生密切相关的突变基因有FLNC、RBM20、TPM、MYH7、ACTC1、DMD、SCN5A、PKP2[62]等。
2.3 ARVC
ARVC 又称右心室发育不良或右室心肌病,是指右心室功能障碍(局部或整体),伴或不伴有左心室疾病;同时还有组织学改变和(或)符合相应标准的心电图异常表现,其特征为右心室心肌进行性被纤维脂肪组织所取代,临床表现为室性早搏、室性心动过速、心室扑动、心室颤动等室性心律失常引起的心悸、头晕、晕厥,甚至引发SCD[80]。其中,23%的患者以猝死为首发症状[81]。ARVC 患者通常在20~50 岁出现与室性心律失常相关的症状,是青壮年及运动员最常见的猝死原因之一[82]。ARVC 是一种常染色体显性遗传疾病,估计患病率为1/5 000~1/2 500,多发生于年轻男性人群,男女性比例约为3∶1[83]。已有超过10 个基因被报道为ARVC 的致病基因,超过一半的ARVC 患者携带桥粒相关基因突变[84]。有研究[58]发现,有6 个(PKP2、DSP、DSG2、DSC2、JUP和TMEM43)具有强有力的证据并且被归类为ARVC 因果关系的确定性基因,可成为ARVC 诊断的主要标准。PKP2是ARVC最常见的致病基因,占所有ARVC 病例的25%[85]。有研究[86]发现,在90 名ARVC 患者中鉴定出57 名突变携带者,其中40 人是PKP2突变携带者,并发现多DPGM突变较单DPGM突变的ARVC 患者临床表现更明显,晕厥及SCD 风险明显更高。根据2015 年欧洲心脏病学会(European Society of Cardiology,ESC)《室性心律失常患者管理和心脏性猝死预防指南》,复合或双基因桥粒突变被列为ARVC 的独立危险因素。在多变量模型分析中,多个桥粒基因突变和男性是主要心律失常事件的独立预测因子,危险比分别为2.3和2.9[87]。有研究[88]报道,双向重测序结果显示所有ARVD5 遗传单倍型中都有跨膜蛋白43(transmembrane protein 43,TMEM431073C→T,S358L)的罕见变异,在对照人群中未发现;TMEM43(1073C→T,S358L)的错义突变可导致完全外显的、受性别影响的ARVD5。其他与HCM 发生密切相关的突变基因有TMEM43、LMNA、PLN、TTN和KCNQ1等[55,63]。
3 死后基因检测技术在法医学的应用和发展
在过去的数十年中,由于测序成本较为昂贵,限制了测序技术在法医病理学实践中的应用。随着医学与分子遗传学研究的发展与不断深入,基因突变被证实与遗传性心脏病的发生和发展密切相关,基因检测成为对此类疾病进行明确诊断的必要手段,死后基因检测技术也逐渐用于法医学鉴定。对不明原因猝死(sudden unexplained death,SUD)者,许多鉴定中心仅对4 个最常见的相关基因(KCNQ1、KCNH2、SCN5A和RYR2)的蛋白质编码外显子进行直接DNA测序[3]。由于基于Sanger 测序的检测仅限于分析特征明确、最普遍的基因,基于人群的研究[3]表明,所谓四基因分子(KCNQ1、KCNH2、SCN5A和RYR2)尸体检验的检出率为15%~20%。这些数据表明,仍有很大部分SCD 病例未被检测到疾病相关基因的变体。出于这个原因,当检测一组与通道病和心肌病相关的基因未能找到可以解释死者表型的变体时,下一代测序(next generation sequencing,NGS)技术也逐渐应用于法医学实践中死亡原因的鉴定工作[89]。NGS 技术能够对相对少量的DNA 样本测序,包括对所有基因的蛋白质编码的整个外显子组或整个基因组进行测序,即全外显子组测序(whole exome sequencing,WES)和全基因组测序(whole genome sequencing,WGS),该技术具有高通量、低成本的优势。因此,NGS 技术允许对所有主要疾病相关基因以及在任何特定疾病中不太常见的基因进行基因检测[90]。最好的方法是技术组合,即对目标疾病相关基因和候选基因进行大规模并行测序,并辅以Sanger 测序对已识别的潜在致病基因进行验证。
不明原因的SCD 病例可能是由潜在的遗传性疾病引起的。部分案例因不伴有心脏结构异常,尸体解剖呈阴性改变,在对其他的非SCD 诱因进行排除之后,只能作出推测性诊断,一般将这些案例判定为青壮年猝死综合征、抑制死或者是死因不明。但出具此类鉴定意见难免会引起质疑。因此,寻找可靠的诊断依据,合理解释死亡原因,成为法医学鉴定亟待解决的问题之一。虽然法医工作者可在尸体检验时通过肉眼或显微镜下观察到死者存在心肌病的组织病理学改变,但也应对所有疑似离子通道病和遗传性心肌病死者进行死后基因检测,并在可能的情况下扩展到所有相关家庭成员。这不仅可以确定死者确切的心肌病发病机制,提高猝死原因的诊断准确性,还可以明确15%~20%不明原因的SCD 病例的死亡原因[91]。一项研究报告[91]表明,与单独的尸体检验相比,尸体检验与基因检测和家庭筛查相结合会有助于发现儿童和年轻人SCD 的死因。所有发生意外猝死的家庭,尤其是当死者基因检测结果显示致病性变异时,可进一步检测和识别其他具有患病高风险的亲属,以确定可能有同样遗传性心脏病风险的亲属[3],可减少死者亲属猝死发生率。通过早期的基因筛查有助于SCD的预防、危险分级与治疗,以减少SCD 的风险。在法医学实践中,对SCD 者进行死后基因检测和对其一级亲属进行心脏遗传学检查,这将在阐明SCD 的根本原因方面提供更大的帮助,并有助于降低家属进一步猝死风险。为了提高不明原因SCD 的诊断率,除了基因检测技术的不断发展外,法医工作者应做到以下几个方面:(1)寻找完整的发病前临床病史,包括晕厥发作史、劳力症状、并发疾病、近期药物治疗、既往心电图等;(2)调查全面的家族史,重点确定心脏病、过早猝死或可疑死亡(如青壮年猝死综合征、婴儿猝死综合征)的家族史;(3)对死者的关键器官(心、脑)进行详细的宏观和组织学评估;(4)为获得最优样本来源,采集5~10 mL 乙二胺四乙酸(ethylenediaminetetraacetic acid,EDTA)抗凝血或冷冻的心、肝或脾组织用于DNA 提取和检测[4];(5)多学科合作,包括检测样本由专业心脏遗传学检测机构进行检测,检测结果由法医病理学家、心脏病学家和临床遗传学家进行准确的整体解释等。
综上所述,将基因检测作为法医传统检验的补充手段,使之成为法医病理学鉴定工作强有力的辅助工具,应用于法医学实践中死亡原因的鉴定工作。未来可通过进一步的实践,总结出类似于离子通道病和遗传性心肌病引发SCD 的案例的诊断依据,制订关于检测到意义未明变体的SCD 病例的管理和解释的具体法医指南,以期更好地应用于法医病理学鉴定实践。
