液相色谱-串联质谱联用定量分析食品中苯嘧磺草胺残留
2022-10-13李丽君郑美娟
袁 晓,李 青,李丽君,郑美娟
(1. 广电计量检测(湖南)有限公司,湖南 长沙 410221;2. 贵州省分析测试研究院,贵州 贵阳 550002)
苯嘧磺草胺是由巴斯夫公司开发的一款脲嘧啶类除草剂。化学名称N'-[2-氯-4-氟-5-(3-甲基-2,6-二氧-4-(三氟甲基)-3,6-二氢-1(2H)-嘧啶)苯甲酰]-N-异丙基-N-甲基硫酰胺,分子式C17H17ClF4N4O5S,分子量500.85[1]。对于草甘磷和乙酰乳酸合成酶(ALS)抑制剂如磺酰脲类、嘧啶水杨酸类有抗性的阔叶杂草和一些莎草科杂草,苯嘧磺草胺有较好的防治效果,其主要作用机制是抑制光合系统中的原卟啉原IX 氧化酶(PPO)活性,造成原卟啉IX(Proto)及过氧化氢在叶片组织中积累,在光照下Proto 分子与氧反应形成单态氧与氧基,使细胞膜的不饱和脂肪酸过氧化,造成膜的透性与功能迅速丧失,导致组织坏死、生长受抑制和植株死亡[2]。
GB 2763—2021《食品安全国家标准 食品中农药最大残留限量》中针对不同的动物和植物源性食品规定了苯嘧磺草胺的临时限量(0.01~1 mg/kg)[3],但是没有指定检测方法。查阅现行有效的农药残留标准,也未找到相关的检测标准。在《关于在各种食品中设立苯嘧磺草胺最大残留限量的合理化意见》(EFSA 2012b)[4]中提到了欧洲食品安全局建立的苯嘧磺草胺检测方法,采用高效液相色谱串联质谱,提取溶剂为甲醇水溶液(70/30, v/v)和乙腈,定量限为0.03 mg/kg(植物源性食品)和0.01 mg/kg(动物源性食品),具体的色谱和质谱条件没有披露。
苯嘧磺草胺是一种新型农药残留,目前,国内针对苯嘧磺草胺的相关研究报道很少,现有的文献报道都是涉及药物合成[5-6]、除草活性测定[7-8]及原药中含量测定[9-10]。原药中苯嘧磺草胺测定方法为液相色谱法,前处理为直接溶解上样,对于复杂的植物源性食品和动物源性食品中低含量苯嘧磺草胺的检测方法还未见报道。检测方法的缺失必然会给农药残留量监测和施用控制带来隐患,本研究旨在建立一种液相色谱串联质谱法测定食品中苯嘧磺草胺残留量的方法,为苯嘧磺草胺的代谢研究和监管提供技术支撑。
1 材料与方法
1.1 仪器设备
高效液相色谱质谱仪:Waters XEVO TQ-S micro(美国沃特世公司);超高速冷冻离心机[力新仪器(上海)有限公司];氮吹仪(上海安谱实验科技有限公司);涡旋振荡器(上海迈旗环保科技有限公司);电子天平(ME204E,梅特勒托利多公司);固相萃取仪(上海屹尧仪器科技发展有限公司);ProElut DPC 固相萃取柱(迪马科技有限公司);0.22 μm 的有机滤膜(迪马科技有限公司)。
乙腈(色谱纯);甲醇(色谱纯);甲酸(99%);氯化钠(分析纯),苯嘧磺草胺标准品,均购自上海安谱科技有限公司。
1.2 试剂与样品
标准储备液:苯嘧磺草胺标准品(PESTANAL®),CAS 号372137-35-4,纯度≥99%。准确称取0.01 g苯嘧磺草胺标准品于10 mL 容量瓶,加入乙腈溶解后定容至刻度。配制成浓度为1 000 μg/mL的标准储备液。于-18℃保存,有效期6 个月。
标准工作液:准确移取0.1 mL 的标准储备液于10 mL 容量瓶,用乙腈定容,配制成浓度为10 μg/mL的标准中间液,于-18℃保存,有效期1 个月。
1.3 样品处理
准确称取2.00 g 样品(猪肉、鸡蛋、上海青、梨、牛奶)于离心管,加入10 mL 乙腈和4.00 g NaCl,混合涡旋1 min,超声提取10 min,6 000 r/min 离心5 min,取5 mL 上清液待净化。加10 mL 乙腈活化固相萃取柱,弃去上清液,将待净化液加入小柱,收集流出液,加入10 mL 乙腈,合并流出液,50 ℃水浴氮吹至近干,用1 mL 乙腈溶剂残留物,过0.22 μm 滤膜后供液相色谱质谱仪测定。
1.4 仪器条件
1.4.1 液相条件液相条件如下:色谱柱为C18 柱(2.6 μm,100 mm×3 mm);流速0.4 mL/min,进样量1.0 μL,柱温35℃,流动相A 相为0.1%甲酸水,B 相为乙腈。梯度洗脱条件见表1。
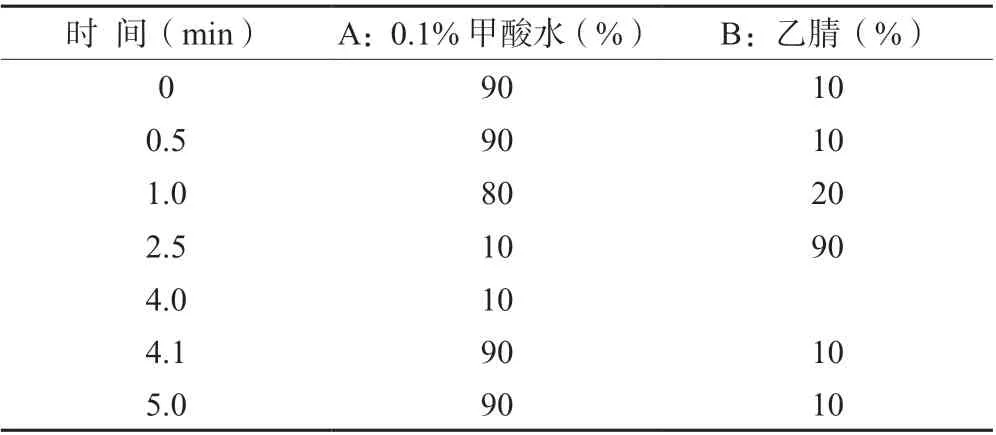
表1 梯度洗脱条件
1.4.2 质谱条件质谱条件如表2 所示。扫描模式MRM 扫描,正负离子同时扫描,气帘气30 Psi,离子化电压正离子+5 500V,负离子-4 500V,去溶剂气温度550℃,喷雾气Gas1:55 Psi,辅助加热气Gas1:55 Psi。
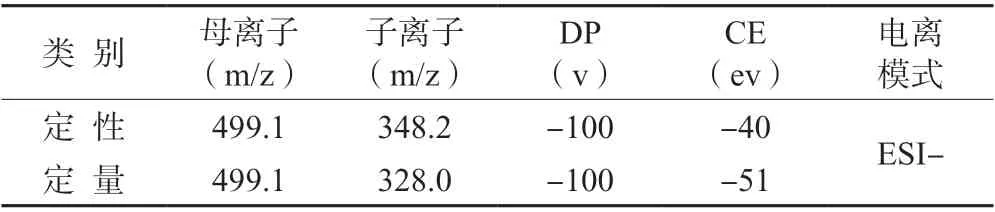
表2 苯嘧磺草胺质谱条件
2 结果与分析
2.1 前处理条件确认
在鸡蛋样品中加入一定浓度的目标物,混合均匀后于4℃放置24 h。称取同等质量的多份上述鸡蛋样品,加入100 μg/kg 苯嘧磺草胺标准物,分别加入5、10、12、15 mL 的乙腈,加入同样质量的NaCl,超声提取净化后进行测定。
表3 是采用不同体积的乙腈提取加标样品后获得的峰面积、响应值和检测值。结果表明,随着提取体积的增加,目标物的检测值依次增加,10 mL 与5 mL相比检测值增加明显,而使用12 mL 和15 mL 溶剂提取后,峰面积与检测值无明显差异,说明10 mL 乙腈的提取效率已可满足前处理的需要,最终选择10 mL 作为提取液体积,不同提取液体积测试结果对比见表3。
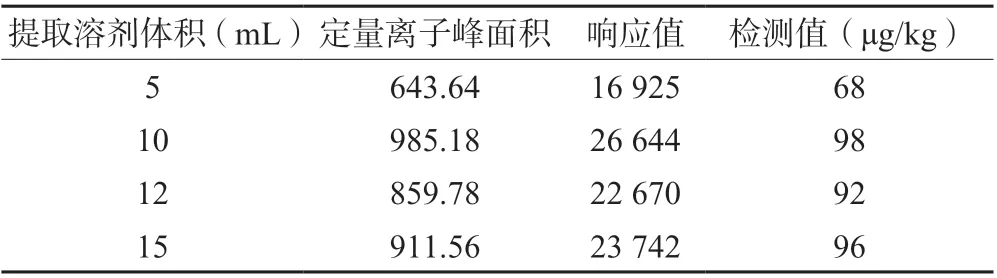
表3 不同体积提取溶剂结果对比
2.2 仪器条件的确定
选择乙腈-水、甲醇-水、甲酸水-乙腈的流动相组合进行比对,主要考察目标物的分离度,同浓度下的响应值,在以乙腈∶0.1%甲酸溶液为流动相时,各目标物的分离度效果最好,最终其作为本方法的流动相。通过对苯嘧磺草胺标准物质注入质谱产生的碎片离子进行分析,发现有348.2 和328 两个较大的离子碎片,且328 碎片响应度高且稳定,可以选作定量离子,将响应低的348.2 碎片作为定性离子。图1 a-c 分别为苯嘧磺草胺定性离子、定量离子、总离子流解析图。
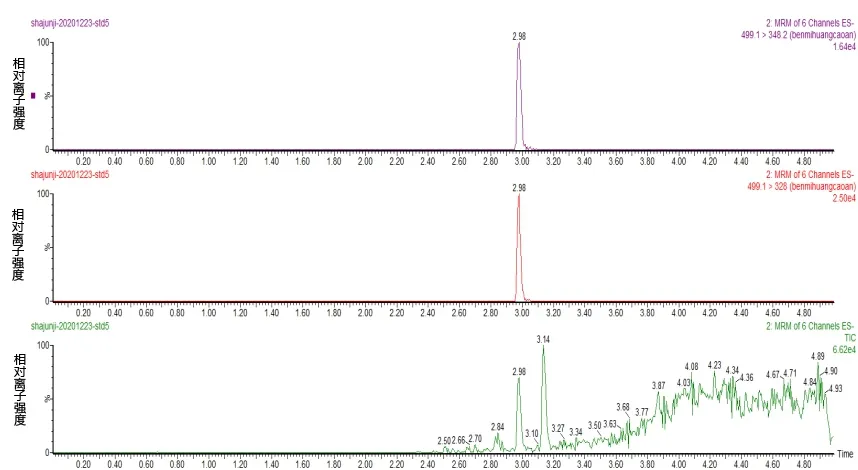
图1 苯嘧磺草胺定量离子(上)、定性离子(中)和总离子流(下)图
2.3 线性范围、标准曲线、检出限及定量限
用标准工作液配制成浓度为10、20、50、100、200 ng/mL 的工作曲线(图2)。以定量离子峰面积为纵坐标,标准物质浓度为横坐标绘制曲线。检出限按信噪比S/N=3,定量限按信噪比S/N=10 计算。在0.01~10.0 mg/kg 范围内苯嘧磺草胺线性关系良好,线性方程为y=24.1361x-16.0385,R=0.9996,检出限为0.002 mg/kg,定量限为0.006 mg/kg。
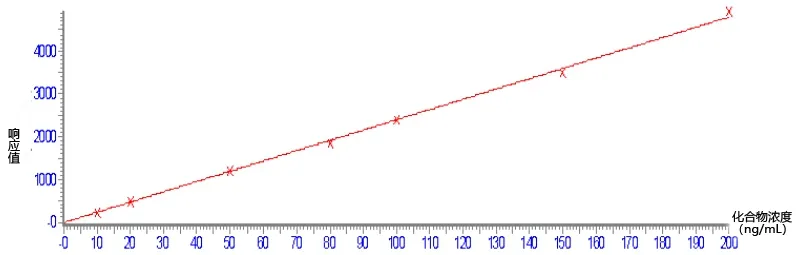
图2 苯嘧磺草胺标准曲线
2.4 不同基质中的响应度
针对常见的食品,选择猪肉、鸡蛋、蔬菜(上海青)、水果(梨)、鸡蛋、牛奶分别配制基质曲线。表4 为不同食物基质中10、20、50、80、100 μg/kg 5 个浓度定量离子峰面积与溶剂基质的对比,由表4 可以看出,苯嘧磺草胺在5 种基质中的响应度与溶剂基质差异不大,虽然牛奶基质中苯嘧磺草胺整体响应度相对较低,但由图3 可知,曲线最低浓度在牛奶基质中也能很好的定性和定量,常见的猪肉、鸡蛋、蔬菜、水果、牛奶中添加10 μg/kg 苯嘧磺草胺质谱图见图4。从图4中可以看出,各基质均能很好的定性和定量。综上所述,此检测方法能适用于上动物源性和植物源性食物中苯嘧磺草胺残留量的检测。
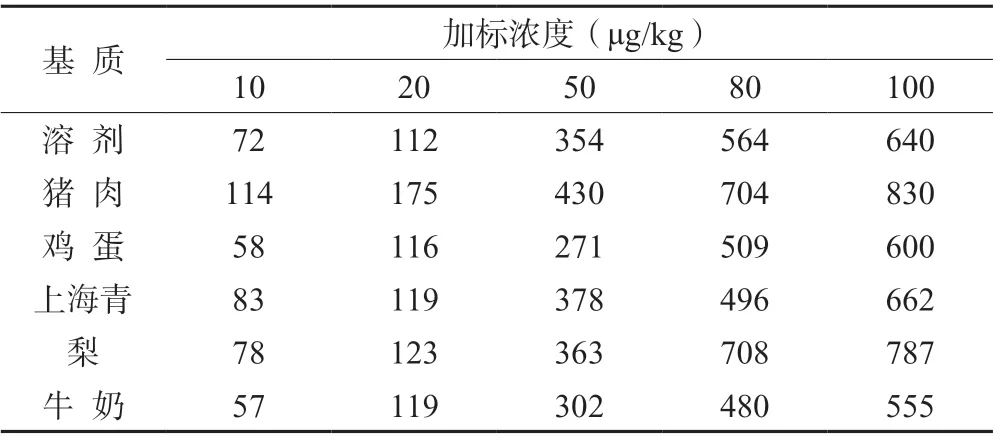
表4 不同基质基中定量离子峰面积对比
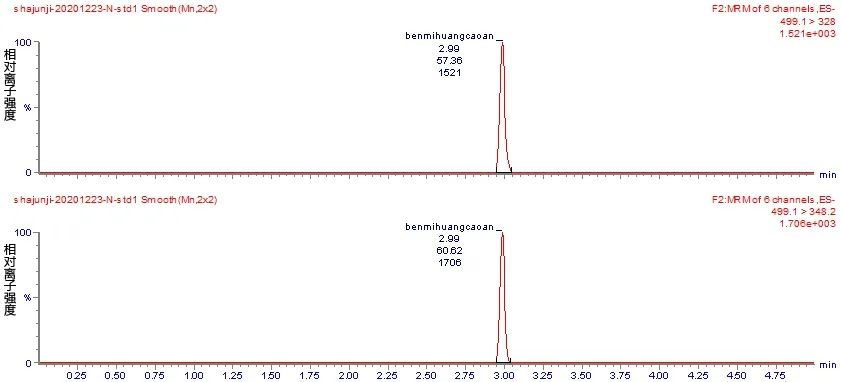
图3 牛奶中添加10 μg/kg 苯嘧磺草胺检测的定量离子(上)、定性离子(下)图
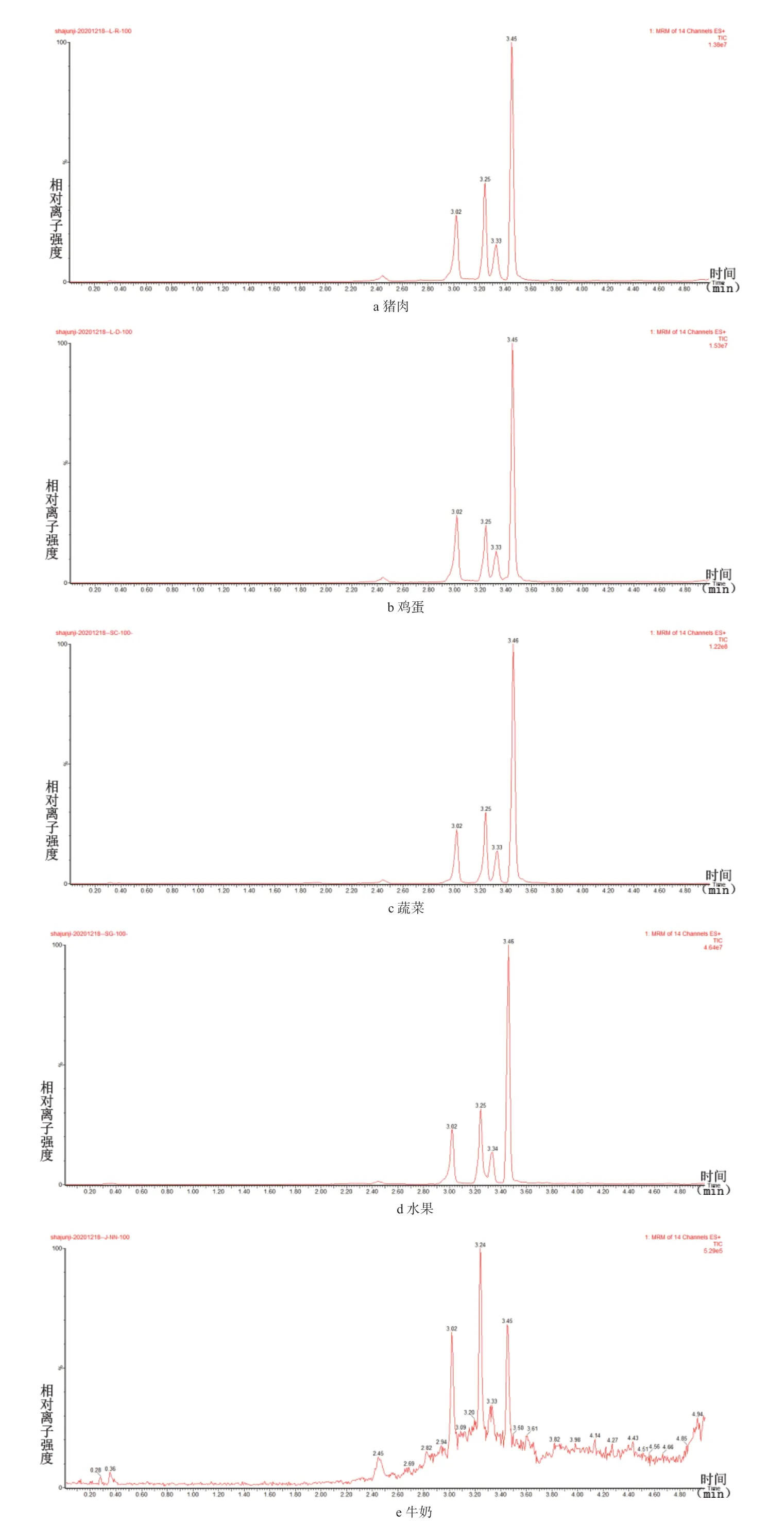
图4 不同基质中添加10 μg/kg 苯嘧磺草胺质谱图
2.5 方法准确度实验
在不同的植物源性食品基质和动物源性食品基质中,分别添加不同浓度的苯嘧磺草胺回收率在88.9%-110.3%之间(表5),符合检测要求。
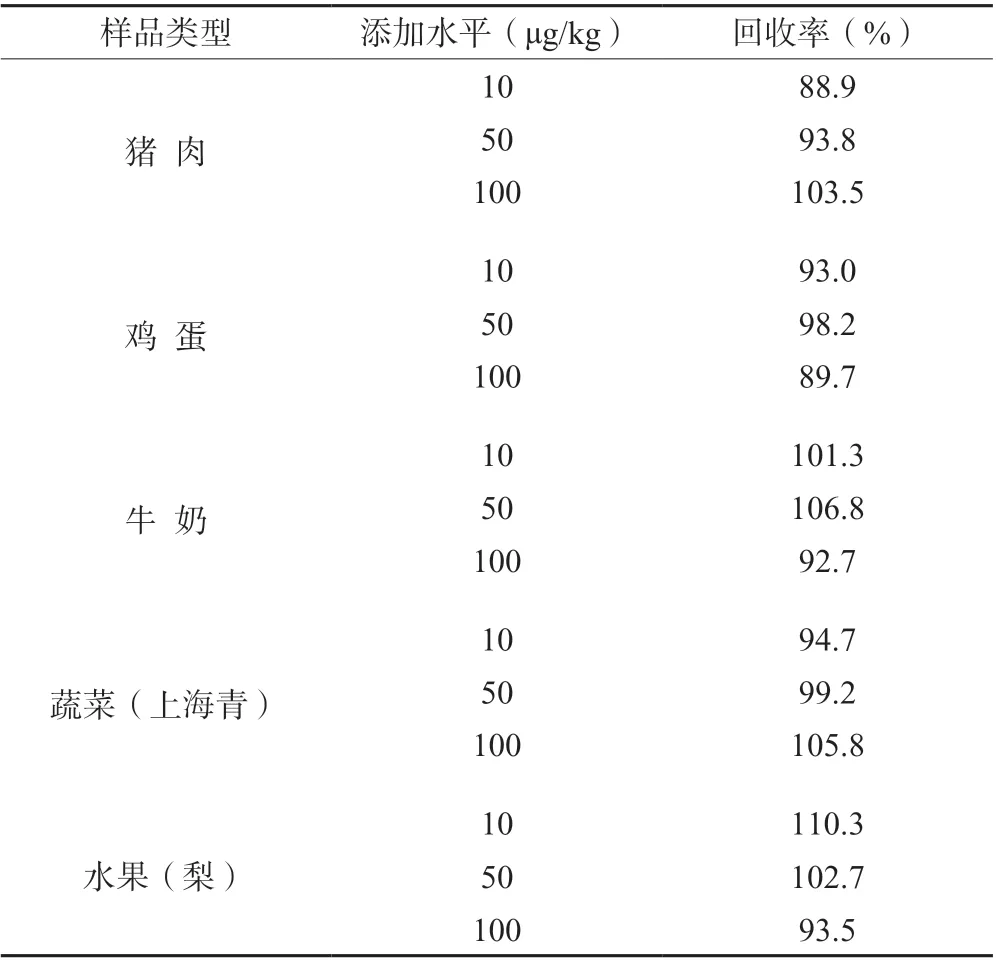
表5 不同基质基中三添加水平回收率对比
3 讨 论
欧洲食品安全局(European Food Safty Authority,EFSA)在2012 年公布的《关于在各种食品中设立苯嘧磺草胺最大残留限量的合理化意见》(EFSA 2012b)[4]中收集了苯嘧磺草胺的急性毒性、短期毒性等毒理学研究数据,同时设计了一系列使用试验(针对作物)、喂养试验(针对牲畜),然后检测苯嘧磺草胺在动植物体内残留,最后评估出苯嘧磺草胺的可接受摄入量为0.05 mg/kg 体重,急性中毒参考剂量也是同样水平。该文件依据评估数据制定了苯嘧磺草胺在不同食品中的残留量限值,同时还指出苯嘧磺草胺的主要代谢物为三氟乙酸(TFA),但是三氟乙酸缺乏毒理学参考数据和残留量数据,无法进行风险评估。因此欧洲食品安全局在2014 年又发布了《不同作物中苯嘧磺草胺限值的合理化建议-基于代谢物三氟乙酸相关风险评估》(EFSA 2014b)[11],公布了三氟乙酸的毒理学研究数据,并对哺乳动物中三氟乙酸暴露风险进行了评估,根据评估数据修改了部分食品中的苯嘧磺草胺残留量限值。
苯嘧磺草胺是名副其实的畅销农药。苯嘧磺草胺在中国的专利为2001 年申请的《尿嘧啶取代的苯基氨磺酰羧酰胺》[13],已经于2021 年4 月到期。脱离专利保护之后,苯嘧磺草胺在中国的产量和应用量有可能会大大增加。值得注意的是,国标中规定某种农药限量为临时限量,一般是因为该物质没有完整毒理学结论、膳食消费数据或没有检验方法,无论是哪一种情况,对于一种正在被日益广泛使用的农药来说,缺少可靠的定量检测方法都是一个极大的隐患。
欧洲食品安全局建立的苯嘧磺草胺检测方法,采用高效液相色谱串联质谱,提取溶剂为甲醇水溶液(70/30, v/v)和乙腈,定量限为0.03 mg/kg(植物源性食品)和0.01 mg/kg(动物源性食品),具体的提取方法、色谱条件和质谱条件没有披露。而此研究的方法定量限优于欧盟方法,提取溶剂也由2 种改为1 种,前处理复杂程度和试验成本应该也有所降低。
4 总 结
研究建立了一种液相色谱-串联质谱测定植物源性食品中苯嘧磺草胺残留量的方法。样品经乙腈超声提取,过固相萃取柱净化,氮吹浓缩后上机检测。在0.01~10.0 mg/kg 范围内线性关系良好,线性方程为y=24.1361x-16.038 5,(R=0.999 6),检出限为0.002 mg/kg,定量限为0.006 mg/kg。方法稳定性较好,精密度高,检出限和定量限能够满足GB 2763—2021 中苯嘧磺草胺的限值要求,能够为苯嘧磺草胺的后续研究以及监测提供技术支撑。
