高效液相色谱串联质谱法检测阿奇霉素原料药及其制剂中6种N-亚硝胺类基因毒性杂质*
2022-10-11郝丽娟徐艳梅高燕霞崔巧利
郝丽娟,徐艳梅,高燕霞△,刘 峰,崔巧利
(1.河北省药品医疗器械检验研究院·仿制药质量控制与评价重点实验室,河北 石家庄 050227;2.石药集团中奇制药技术<石家庄>有限公司·河北省制技工程技术研究中心·新型药物制剂与辅料国家重点实验室,河北 石家庄 050035)
阿奇霉素是首个十五元环大环内酯类抗菌药物,化学名为9a-甲基-9-脱氧-9a-氮杂-9a-同红霉素A,由红霉素A经过肟化、贝克曼重排、还原、甲基化等一系列反应制备而成[1-2],主要用于治疗呼吸道、软组织、泌尿系统疾病及皮肤感染等,疗程短,副作用少[3]。基因毒性杂质是可以造成基因突变或体内诱变的物质,痕量即可引起癌变、畸变[4]。缬沙坦、雷尼替丁、二甲双胍中先后检出N-亚硝胺类基因毒性杂质,《世界卫生组织国际癌症研究机构致癌物清单》已将多个N-亚硝胺类化合物归为A类致癌物质。其产生的原因可能与工艺过程、降解途径、污染引入等有关[5-6]。阿奇霉素的11位糖环上存在二甲胺基结构,是典型基因毒性杂质的警示结构,且原料药的合成过程存在亚胺类中间体,易通过氧化等途径引入基因毒性杂质[7-8]。起始原料红霉素A和阿奇霉素注射剂中均检出亚硝胺类基因毒性杂质N-亚硝基二甲胺(NDMA)[9-10]。我国为全球重要生产国和出口国,国家药品监督管理局网站公布的600多种阿奇霉素药物的注册申报和出口都需要对其N-亚硝胺类杂质进行研究,但目前未见关于检测阿奇霉素药物中N-亚硝胺类杂质的相关研究。参照各国药品监管机构发布的N-亚硝胺类基因毒性杂质的通用检查方法[11]并参考文献[12-14],本研究中建立了测定阿奇霉素原料药及其制剂中6种N-亚硝胺类基因毒性杂质的高效液相色谱串联质谱(HPLCMS/MS)法,方法灵敏度高、结果准确,为有效控制阿奇霉素及其制剂中的基因毒性杂质提供了依据。现报道如下。
1 仪器与试药
1.1 仪器
岛津LC-30AD型高效液相色谱仪(日本岛津公司);SCIEX ATRAP 6500型质谱仪(美国AB公司);XS105型电子天平(瑞士Mettler Toled公司,精度为十万分之一);Milli-Q型纯水制造系统(美国Millipore公司)。
1.2 试药
NDMA对照品(TMstandard公司,批号为1681903,含量以99.9%计);N-亚硝基-N-甲基-4-氨基丁酸(NMBA)对照品(批号为51-KFA-33-1,含量以100%计),N-亚硝基二乙胺(NDEA)对照品(批号为50-GHZ-66-1,含量以100%计),均购自加拿大TRC公司;N-亚硝基乙基异丙胺(NEIPA)对照品(批号为D10252828,含量以100%计),N-亚硝基二异丙胺(NDIPA)对照品(批号为D10252827,含量以100%计),N-亚硝基二丁胺(NDBA)对照品(批号为D10252829,含量以100%计),均购自HIC公司;甲醇(色谱纯,德国默克股份有限公司);水为超纯水。抽检样品:阿奇霉素(原料药,批号分别为108200513,108200514,108200518);阿奇霉素肠溶片(批号为FAR2901005);阿奇霉素胶囊(批号为21012001);阿奇霉素干混悬剂(批号为207210201)。
2 方法与结果
2.1 色谱与质谱条件
2.1.1 色谱条件
色谱柱:ACE Excel 3 C18-AR柱(150 mm×4.6 mm,5 μm);流动相:0.1%甲酸水溶液(A)-甲醇(B),梯度洗脱(洗脱程序见表1);流速:0.6 mL/min;柱温:40℃;进样量:10 μL。
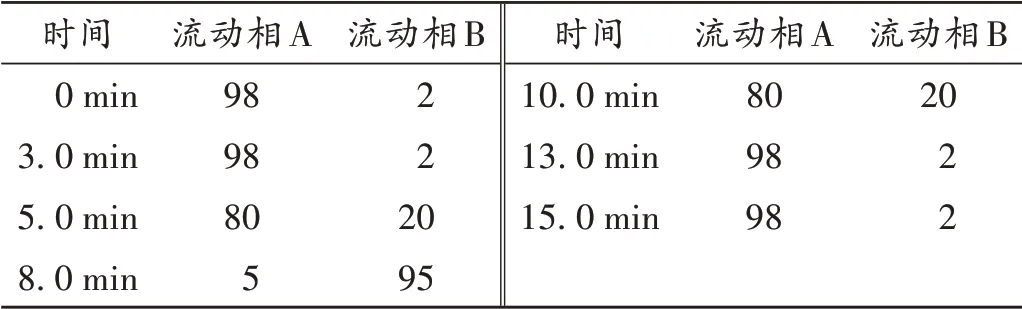
表1 流动相梯度洗脱程序(%)Tab.1 Program of gradient elution of mobile phase(%)
2.1.2 质谱条件
电离模式:正离子;离子源:大气压化学电离(APCI)源;监测模式:多反应监测(MRM);中性(NC)电流:3 mA;气帘气(CUR):206.8 kPa;雾化气(GS1):241.3 kPa;离子源温度:350℃。6种N-亚硝胺类基因毒性杂质的保留时间及质谱分析参数见表2。
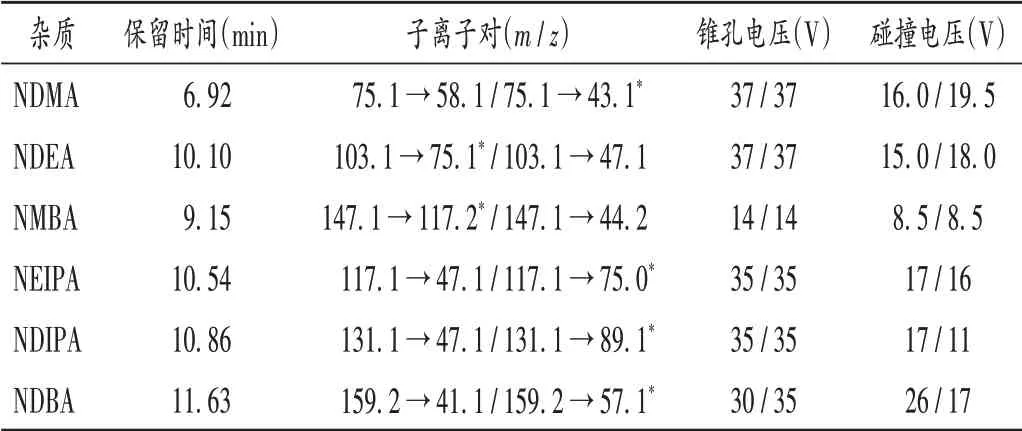
表2 6种N-亚硝胺类基因毒性杂质的保留时间及质谱分析参数Tab.2 Retention time and HPLC-MS/MS parameters of the six N-nitrosamine genotoxic impurities
2.2 溶液制备
混合对照品溶液:分别取6种N-亚硝胺类基因毒性杂质对照品适量,精密称定,加水稀释并定容,制成NDMA,NMBA和NDEA,NEIPA,NDIPA,NDBA的质量浓度分别为265 μg/mL和960 μg/mL的混合对照品贮备液。取上述混合对照品贮备液各适量,用水逐级稀释制得系列质量浓度的混合对照品溶液(NDMA和NMBA的系列质量浓度分别为48.00,24.00,4.80,2.40,0.96,0.48 ng/mL,NDEA,NEIPA,NDIPA,NDBA的系列质量浓度分别为13.25,6.62,1.33,0.66,0.26,0.13 ng/mL)。
供试品溶液:取原料药0.5 g,精密称定,置10 mL容量瓶中,加水5 mL,涡旋5 min,用水稀释并定容,振摇10 min,以4500 r/min的速率离心10 min,制成质量浓度为50 mg/mL的溶液,滤过,取续滤液,即得原料药供试品溶液。取样品(或内容物)20片(或粒、袋)研细,混匀,取细粉适量(约相当于阿奇霉素0.5 g),精密称定,置10 mL容量瓶中,加水5 mL,涡旋5 min,用甲醇稀释并定容,振摇10 min,以4500 r/min的速率离心10 min,滤过,制成质量浓度为50 mg/mL的溶液,取续滤液,即得制剂供试品溶液。
2.3 方法学考察
专属性试验:取空白溶剂及2.2项下混合对照品溶液、原料药供试品溶液、制剂供试品溶液,按2.1项下条件进样测定。结果6种N-亚硝胺类基因毒性杂质均无干扰峰,表明方法专属性良好。色谱图见图1。
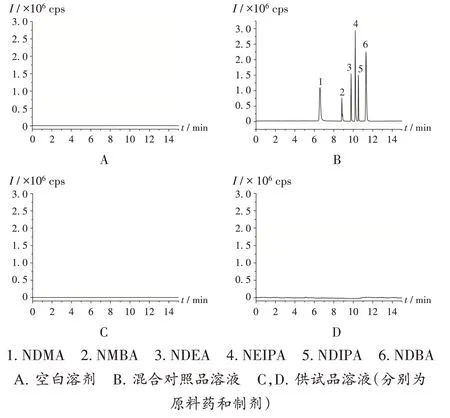
图1 专属性试验高效液相色谱串联质谱图1.NDMA 2.NMBA 3.NDEA 4.NEIPA 5.NDIPA 6.NDBAA.Blank solvent B.Mix reference solution C,D.Test solution(azithromycin API and its preparations,respectively)Fig.1 HPLC-MS/MS chromatograms of the specificity test
线性关系考察、检测限和定量限确定:取2.2项下系列质量浓度的混合对照品溶液适量,按2.1项下色谱与质谱条件分别进样测定,记录色谱图。以待测成分质量浓度(X,μg/mL)为横坐标、峰面积(Y)为纵坐标进行线性回归。结果表明,NDMA,NMBA和NDEA,NEIPA,NDIPA,NDBA质量浓度分别在0.48~48.00 ng/mL和0.13~13.25 ng/mL范围内与峰面积线性关系良好。分别以混合对照品溶液信噪比(S/N)为3∶1和10∶1的质量浓度为检测限与定量限。结果见表3。
精密度试验:精密量取2.2项下混合对照品溶液适量,按2.1项下色谱与质谱条件平行测定6次。结果见表3,表明仪器精密度良好。
稳定性试验:精密量取2.2项下系列质量浓度的混合对照品溶液,分别于室温放置0,1,2,3,6,10 h时按2.1项下色谱与质谱条件进样测定。结果见表3,表明对照品溶液在室温下放置10 h稳定性良好。
重复性试验:取原料药样品(批号为108200513),依法平行制备供试品溶液6份,分别量取对照品溶液适量,同法制备加标供试品溶液,按2.1项下色谱与质谱条件进样测定。结果见表3,表明方法重复性良好。
加样回收试验:取原料药样品(批号为108200513),按2.2项下方法制备供试品溶液,共9份,分别精密加入2.2项下混合对照品溶液适量,制备成高、中、低3个质量浓度,各3份,按2.1项下色谱与质谱条件进样测定,并按外标法计算加样回收率。结果见表3,表明方法准确度良好。
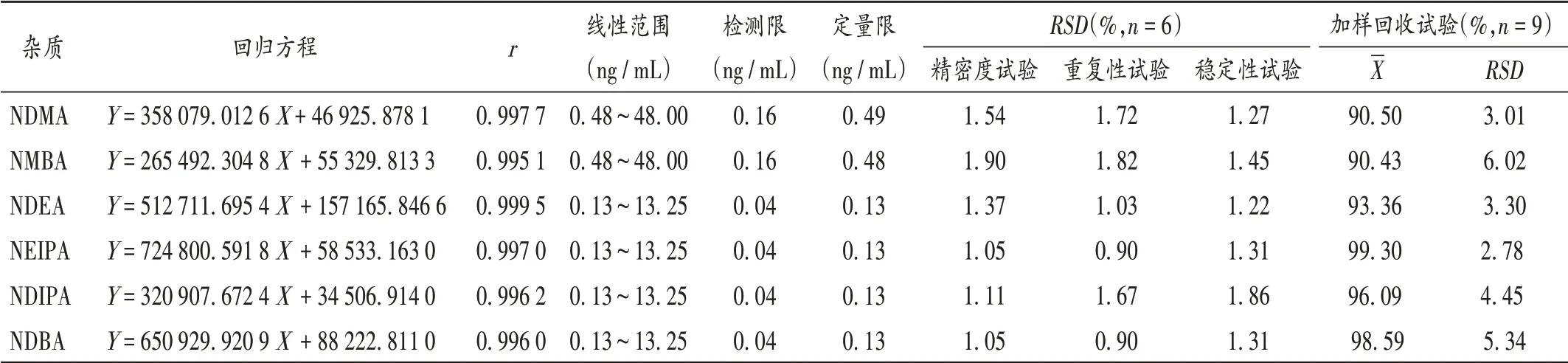
表3 方法学考察结果Tab.3 Results of the methodological investigation
耐用性试验:更换流动相流速(0.4,0.8 mL/min),起始柱温(35,45℃),流动相B起始比例(0,4%)、不同品牌色谱柱进行测定。结果显示,小范围的改变测定条件,色谱峰峰面积变化不大,表明方法耐用性良好。
强制降解试验:取原料药样品(批号为108200513)4份,按2.2项下方法制备供试品溶液,分别进行强酸、强碱、氧化和加热(乙腈溶解,沸水浴)破坏试验,按2.1项下色谱与质谱条件进样测定,结果降解产物中均未检出6种N-亚硝胺类基因毒性杂质。色谱图见图2。
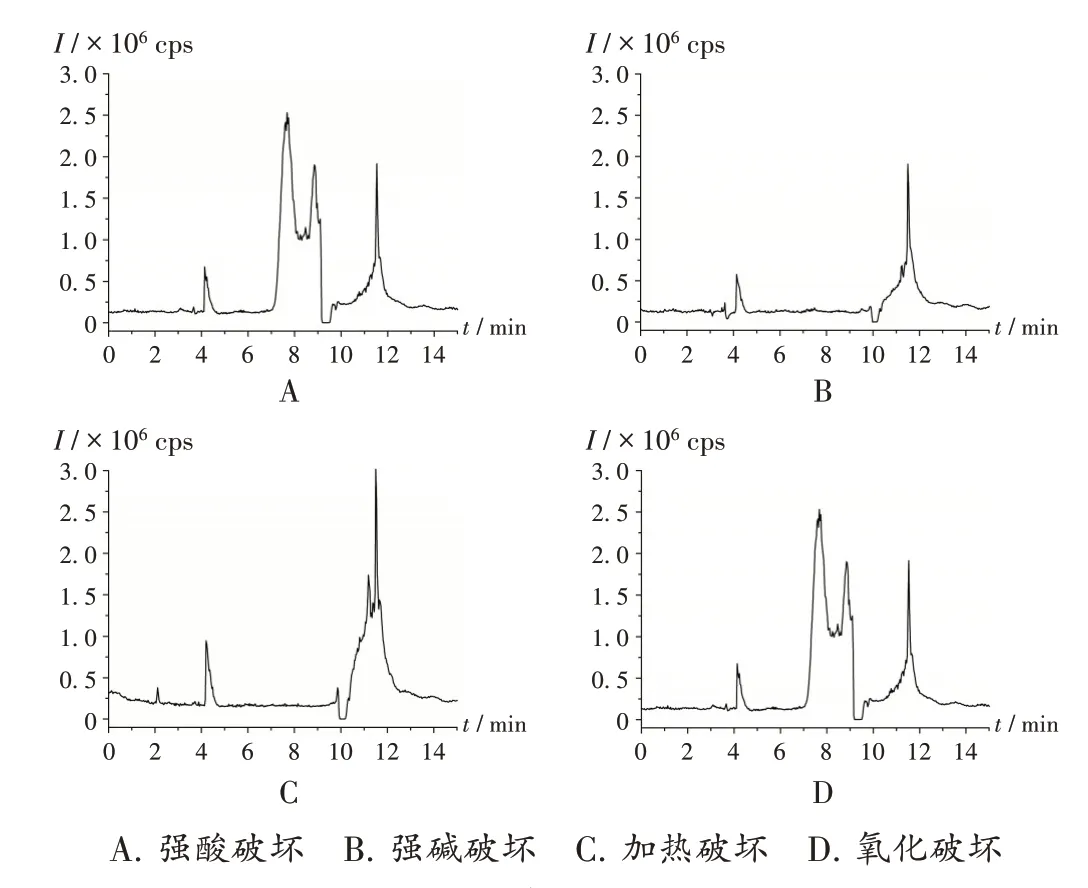
图2 强制降解试验高效液相色谱串联质谱图A.Strong acid damage B.Strong alkali damage C.Heating damage D.Oxidation damageFig.2 HPLC-MS/MS chromatograms of the forced degradation test
2.4 样品检测
分别取3批(批号分别为108200513,108200514,108200518)阿奇霉素原料药和3批(批号分别为FAR2901005,21012001,207210201)阿奇霉素制剂,依法制备供试品溶液,按2.1项下色谱与质谱条件进样测定。结果3批阿奇霉素原料药和3批制剂中均未检出6种N-亚硝胺类基因毒性杂质。
3 讨论
3.1 提取方法选择
以各成分的提取率为指标,考察了甲醇、乙醇、水、50%甲醇等提取溶剂及前处理方法,最终选择水作为溶剂,前处理方法采用先涡旋后振摇、再离心,以提高各成分的回收率。
3.2 色谱条件选择
考察了甲醇-0.1%甲酸水溶液、0.1%甲酸甲醇-0.1%甲酸水溶液、甲醇-0.2%甲酸水溶液、乙腈-0.1%甲酸水溶液等流动相系统,结果以甲醇-0.1%甲酸水溶液为流动相时,各成分色谱峰峰形及分离度均良好,12 min内可完全洗脱。
考察了Waters XBridge C18柱、Agilent Poroshell 120 EC-C18柱、XSelect HSS T3柱、ACE Excel 3 C18-AR柱等色谱柱,结果以ACE Excel 3 C18-AR柱为色谱柱时,各成分色谱峰峰形良好,分离度较高。又通过对柱温及梯度洗脱程序的调整,进一步提高了分离效率,最终确定2.1.1项下色谱条件。
3.3 方法评价
阿奇霉素与敏感微生物的50s核糖体的亚单位结合后,对其蛋白质的合成产生干扰,体外试验和临床研究均表明,阿奇霉素对多种革兰阳性菌、革兰阴性菌需氧致病菌有良好的抑菌效果,临床用途广泛。其分子中含有伯胺、仲胺结构,具有典型基因毒性致癌物的警示结构,在制备和储藏过程中易产生N-亚硝胺类杂质。本研究中建立了测定阿奇霉素原料药及其制剂中6种N-亚硝胺类基因毒性杂质的HPLC-MS/MS法,强制降解产物中均未检出这6种杂质,推测此类基因毒性杂质为工艺杂质,应对原料生产的各个环节和步骤进行严格控制。方法学考察结果表明,所建立的方法操作简便、灵敏度高、结果准确、重复性好,可用于生产企业及药监系统日常检查,监测阿奇霉素原料药及其制剂中的N-亚硝胺类基因毒性杂质。
