肺炎克雷伯菌对多黏菌素的耐药机制研究进展Δ
2022-10-10重庆医科大学附属第二医院药学部重庆400010
徐 特,钱 妍,李 頔(重庆医科大学附属第二医院药学部,重庆 400010)
肺炎克雷伯菌是肠杆菌科克雷伯菌属中最主要的一种病原菌,广泛存在于自然环境中,同时其也可定植于人的肠道、口腔以及皮肤等部位。肺炎克雷伯菌作为院内及社区感染的常见致病菌,可导致肺炎、尿路感染、脓毒血症及肝脓肿等疾病。由于抗生素的大量使用,肺炎克雷伯菌呈现出多药耐药(multiple drug resistance,MDR)趋势,尤其是耐碳青霉烯类肺炎克雷伯菌(carbapenemresistantKlebsiella pneumoniae,CRKP)的出现,意味着可用于治疗肺炎克雷伯菌感染的抗生素越来越少,严重威胁着人类健康[1]。
多黏菌素是由多黏芽孢杆菌合成的一类具有抗菌活性的物质,对肺炎克雷伯菌、铜绿假单胞菌、大肠埃希菌等有着良好的杀菌作用。由于多黏菌素具有神经及肾脏毒性,所以该药在临床上已很少使用。鉴于CRKP严峻的耐药现状以及可使用的抗生素屈指可数,多黏菌素以其独特的抗菌机制成为了治疗CRKP感染的最后一道防线[2]。但随着近数十年来多黏菌素使用的增加,国内外肺炎克雷伯菌耐药株的报道也呈上升趋势[3]。这不仅大大增加了感染的防控成本,也危害着患者的生命安全。基于此,本文就目前已报道的肺炎克雷伯菌对多黏菌素的耐药机制作一综述,以期为多黏菌素的合理使用及耐药肺炎克雷伯菌的防治提供依据。
1 多黏菌素的抗菌机制
多黏菌素是一类环状肽抗菌剂,按结构差异可将此类药物分为A、B、C、D、E类。临床上最常用的是多黏菌素B和多黏菌素E(也称黏菌素)。这2种药物均表现出亲水、亲脂的两性分子特点,只是在肽环上的氨基酸不同——多黏菌素B为D-苯丙氨酸,多黏菌素E为D-亮氨酸(具体化学结构见图1)。多黏菌素B、E对病原菌的作用机制类似,两者化学结构中所含的二氨基丁酸残基可与革兰氏阴性菌外膜脂多糖(lipopolysaccharide,LPS)部件脂质A上的游离磷酸基团直接结合,从而破坏细菌外膜的稳定性,导致菌体破裂、溶解,继而发挥杀菌作用[4-6]。除上述作用机制外,目前还有很多关于多黏菌素杀菌机制的假说,包括影响细菌的结构、呼吸作用、核糖体结合和分裂以及诱导活性氧(reactive oxygen species,ROS)的产生等[7]。
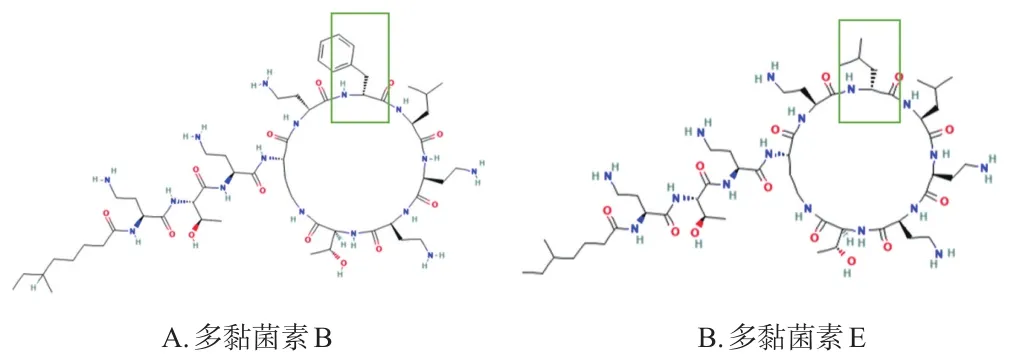
图1 多黏菌素B、E的化学结构式
2 多黏菌素的耐药机制
由于多黏菌素的主要作用靶点是LPS,因此目前大多数多黏菌素耐药机制研究主要集中在LPS的结构修饰上[3]。此外,荚膜多糖过表达、外排泵过表达等机制也被证实与耐多黏菌素肺炎克雷伯菌的出现有关[8-9]。基于此,本文对这几种耐药机制进行归纳介绍。
2.1 LPS的结构修饰
2.1.1 染色体介导的LPS结构修饰 肺炎克雷伯菌的LPS结构修饰主要是向脂质A添加4-氨基-L-阿拉伯糖(4-amino-4-deoxy-L-arabinose,L-Ara4N)或磷酸乙醇胺(phosphoethanolamine,pEtN),这2种修饰会导致LPS结构和电位发生改变,从而减少多黏菌素与肺炎克雷伯菌的结合[10-11]。
上述2种LPS结构修饰与PmrA/B以及PhoP/Q双组分信号转导系统激活密切相关。PmrA/B与PhoP/Q均为跨膜信号转导元件,其中PhoQ和PmrB为组氨酸激酶,可感应外界刺激,从而活化细胞质内的反应调控因子[12]。PhoQ、PmrA、PmrB基因的错义突变导致PmrA/B的活化,从而上调PmrCAB和arnBCADTEF(也称为PmrHFIJKLM)操纵子,进而促进pEtN和L-Ara4N的合成以及向脂质A的转移。其中,pEtN可升高脂质A的电位,L-Ara4N可直接中和脂质A的电荷[13-15]。另外,PhoP/Q双组分信号转导系统的负反馈调节因子MgrB基因的突变也被证实可导致细菌对多黏菌素耐药,其原因一方面在于MgrB对PhoP/Q双组分信号转导系统的抑制作用减弱,导致eptE(pEtN转移酶)促使pEtN向外膜3-脱氧-D-甘露糖醛酸残基转移,另一方面MgrB的插入失活(如可移动元件ISKpn14序列)可活化PmrA调控因子,进而促进L-Ara4N的合成以及向脂质A的转移[16-17]。PmrD作为一类连接PmrA/B及PhoP/Q的信号蛋白,可以阻断经PmrB活化后的磷酸化PmrA的固有去磷酸化,从而促进PmrA下游基因的转录与表达,最终加强LPS结构修饰,导致耐药[18]。
CrrA/B是一类新发现的可能与多黏菌素耐药机制有关的双组分系统,同样由感受器和调控因子组成,其所介导的多黏菌素耐药机制表现为调节CrrB基因临近的H239-3059基因、H239-3062基因(即CrrC基因,与糖基转移相关)来影响LPS修饰[19]。Cheng等[11]研究发现,CrrB的氨基酸替换会降低肺炎克雷伯菌对多黏菌素的敏感性,这是因为CrrB基因的突变可诱导H239-3062(即CrrC)表达,而CrrC可通过PmrA/B促进PmrC和PmrHFIJKLM操纵子的表达,进而使细菌对多黏菌素耐药。
综上,肺炎克雷伯菌对多黏菌素耐药的调控通路主要包括PhoP/Q、PmrA/B、CrrA/B这3类双组分系统,且三者之间存在密切联系(三者参与LPS结构修饰的机制见图2)。

图2 双组分系统参与LPS修饰的机制
2.1.2 质粒介导的LPS修饰 除上述集中由染色体介导的耐药调控基因突变外,革兰氏阴性菌还可通过质粒介导的黏菌素可移动耐药(mobile colistin resistance,mcr)基因及其变异体在菌群间的水平传播而获得耐药[20]。在Du等[21]第一次发现携带mcr-1基因的耐多黏菌素肺炎克雷伯菌后,在中国[22]、意大利[23]、埃及[24]等地已有多篇有关mcr基因及其变异体导致细菌对多黏菌素耐药的报道,且来源包括了人、动物以及环境样本等。mcr基因的独特遗传结构在很大程度上造成了该基因的全球扩散。
mcr基因多位于质粒(包括IncI2、IncHI2、IncX4、IncN等)上,且基因结构中多包含有移动元件ISApl1。mcr基因具有可传播性,含有与以上几类质粒遗传结构类似的细菌更易于通过质粒结合获得耐药性,这也导致了耐药菌株的快速扩散[25]。除mcr-1外,在肺炎克雷伯菌中也发现了mcr-1的等位基因,如mcr-2、mcr-3、mcr-7、mcr-8、mcr-9等[24,26-28]。以mcr-1为例,其编码的pEtN转移酶可催化已合成的pEtN向脂质A磷酸基团转移,增加脂质A位点的阳离子电荷,从而减少多黏菌素与LPS的结合,进而导致细菌对多黏菌素耐药[5]。
2.2 荚膜多糖的过表达
Campos等[29]研究发现,当肺炎克雷伯菌暴露于多黏菌素时,荚膜多糖的过表达可对肺炎克雷伯菌起保护作用。荚膜多糖存在于细菌的最外层,作为物理屏障其可减轻菌体受到的外界伤害。荚膜多糖可通过与多黏菌素结合来减少多黏菌素与LPS的作用。另外,该学者团队还发现,部分肺炎克雷伯菌缺乏含O抗原的LPS,其荚膜多糖是唯一可阻碍多黏菌素作用的屏障。相关研究也发现,肺炎克雷伯菌可从菌体表面释放带负电荷的荚膜多糖,这类荚膜多糖可捕获多黏菌素,使多黏菌素与非活性位点的作用增加,进而减少多黏菌素与细胞外膜的接触,从而影响其正常杀菌作用[30]。Fresno等[8]提出,荚膜多糖是以离子相互作用的形式稳定结合于LPS,当环境中存在多黏菌素时,该离子相互作用会被多黏菌素所带的电荷扰乱,故荚膜多糖可脱离LPS与多黏菌素结合,进而减轻多黏菌素对肺炎克雷伯菌外膜的破坏。另一项关于抗菌肽对肺炎克雷伯菌外膜作用的研究发现,外膜蛋白OmpA缺失的菌株,其荚膜多糖表达下降,进而导致肺炎克雷伯菌对多黏菌素B的敏感性增加[31]。由此可知,可通过抑制OmpA来缓解耐药,这为克服细菌耐药性提供了新思路。
2.3 外排泵的过表达
细菌多药外排泵被认为与耐药密切相关。Srinivasan等[32]研究发现,KpnEF外排泵可介导肺炎克雷伯菌对多黏菌素耐药,该外排泵属于小多药外排家族(small MDR family,SMR),另外KpnEF跨膜转运蛋白失活还可导致荚膜多糖的合成障碍。全基因组测序发现,存在KpnEF基因点突变且同时合并PmrA、CrrB基因突变的肺炎克雷伯菌可对多黏菌素表现出耐药[33]。Padilla等[34]研究发现,当编码acrAB多药外排泵的基因被敲除后,肺炎克雷伯菌对多黏菌素的敏感性相较于野生型菌株发生改变。该外排泵由acrRAB操纵子编码,其中acrA编码锚定于细胞内膜上连接内膜与外膜的间质蛋白,acrB编码锚定于细胞质膜的整合蛋白,acrR编码外排泵的抑制因子。相较于野生型菌株,acrB基因敲除菌株对多黏菌素更为敏感,而acrR敲除的菌株则相反,这说明acrR对药物外排表现为抑制效应。另有研究发现,CrrB基因的点突变可导致属于耐药结节分化(resistance nodulation division,RND)家族的新型外排泵H239-3064的表达减少,当该外排泵过表达时,肺炎克雷伯菌对多黏菌素的敏感性大大降低[35]。相关研究发现,多药外排泵KeXD与H239-3064有着100%的氨基酸一致性,提示KeXD也可因CrrB的突变而出现过表达,从而导致细菌对多黏菌素耐药[36]。RamA和SoxS是AcrBTolC外排泵的调节因子,相较于多黏菌素敏感株,两者在耐药株中的表达更高,其中RamA同时影响着脂质A的合成,在其负调节因子RamR失活的情况下,脂质A的合成增加,因而使细菌表现出对多黏菌素不敏感[37-38]。
3 异质性耐药
由单一分离菌株培养所得的细菌群体中,可能存在不同亚群耐药情况不同的现象,如部分敏感、部分耐药,此种情况为异质性耐药,即有着相同或相似基因型的菌株却表现出不同的耐药表型。一旦暴露于抗生素中,异质性耐药菌株即可被筛选出来[39]。目前,临床常以最低抑菌浓度(minimum inhibitory concentration,MIC)来判断细菌是否耐药,但在某些敏感菌株中,仅以MIC判断则难以排除异质性耐药。虽然,导致异质性耐药发生的机制尚不明确,但已有报道证明,亚群耐药性的出现与增强主要是由包括典型抗性基因在内的基因自发性扩增所致[40]。多黏菌素异质性耐药的出现,很有可能部分与多黏菌素耐药调控相关基因发生不稳定突变有关。Jayol等[41]和Halaby等[42]的研究均发现,多黏菌素异质性耐药的肺炎克雷伯菌的PhoQ基因存在突变,且后者还发现mgrB、yciM基因(编码LPS调节蛋白)和lpxM基因亦存在突变,这提示除LPS结构修饰外,多黏菌素耐药可能还存在其他机制[43]。Cheong等[44]在一株分离自住院患者的肺炎克雷伯菌中发现了mutS的无义突变,该突变直接导致肺炎克雷伯菌对阿米卡星的耐药以及对多黏菌素的异质性耐药,且该基因编码一种广泛存在于肺炎克雷伯菌中的DNA修复酶,这意味此耐药突变很有可能影响肺炎克雷伯菌在临床上的防治过程。
4 结语
多黏菌素作为目前用于治疗多药耐药革兰氏阴性菌感染的“最后防线”,由于其使用增加以及耐药基因的水平传播等,导致肺炎克雷伯菌对其耐药增加。综上所述,肺炎克雷伯菌耐多黏菌素的机制包括细菌外膜LPS的结构修饰(主要为L-Ara4N和pEtN向脂质A转移)、荚膜多糖的过表达、多药外排泵的过表达等。就LPS结构修饰而言,染色体介导的LPS结构修饰直接导致了细菌的获得性耐药,质粒介导的LPS结构修饰则引发了耐药性的水平传播。虽然肺炎克雷伯菌耐药机制可分为上述几种,但在不同的肺炎克雷伯菌菌株中,可能存在多种耐药机制共存的现象,而且肺炎克雷伯菌作为院内感染的主要病原菌,常与其他致病菌共同检出,这也有可能导致产生某些本不属于肺炎克雷伯菌的耐药机制。现有研究可能还无法解释某些临床中存在的问题,如异质性耐药的具体机制等,但相信未来的科学研究将进一步明确肺炎克雷伯菌的耐药机制,为耐药菌的检测以及感染的防控提供证据支撑。虽然目前对已发生的多黏菌素耐药尚无较好的阻断实践,但为避免多黏菌素耐药现状的进一步恶化,应合理优化临床用药,加强监督管理,尽可能减少耐药基因的扩散。
