超高效液相色谱法测定罗汉果中7种成分含量*
2022-10-09彭可垄彭晴菲刘林慧尹春萍
彭可垄,彭晴菲,刘林慧,尹春萍
(1.重庆药友制药有限责任公司,重庆 401121;2.华中科技大学同济医学院药学院,武汉 430030)
罗汉果为葫芦科植物罗汉果Siraitiagrosuenorii(Swingle)的干燥果实,性甘,凉,归肺、大肠经,功能为清热润肺,利咽开音,滑肠通便,用于肺热燥咳,咽痛失音,肠燥便秘[1]。目前,国家药品监督管理局批准的罗汉果相关药品文号数量有100多个,应用广泛。罗汉果中葫芦素类化合物具有抗肿瘤、抗炎、调节免疫、保肝、降血糖以及抗心肌肥大等多种药理作用,主要用于治疗肿瘤、肝脏疾病、炎症、糖尿病和心血管疾病等[2-5]。此外,罗汉果皂苷Ⅴ还可作为难溶性药物的载体,起到增溶作用[6]。研究表明[7],除罗汉果皂苷Ⅴ外,罗汉果苷Ⅱe、罗汉果苷Ⅲe、罗汉果苷Ⅳe、11-O-罗汉果苷Ⅱe和罗汉果苷Ⅲ等化学成分有望成为罗汉果质量标志物(quality marker,Q-Marker)。罗汉果现行质量标准为《中华人民共和国药典》2020年版一部收载的标准,含量测定中仅对罗汉果皂苷Ⅴ进行控制,未对其他罗汉果苷类物质进行控制。YAN等[8]报道一种采用飞行时间质谱法测定5种罗汉果苷成分的方法,但该方法采用飞行时间质谱进行含量测定,不适合作为常规的控制手段。为了能对罗汉果的其他成分进行准确测定,进而为提高药品质量标准奠定基础,需要一个可以同时测定罗汉果中多种成分的分析方法。高效液相色谱法特别是超高效液相色谱法(ultra performance liquid chromatography,UPLC)由于具有柱效高,分离速度快的优势,在中药含量检测中的应用越来越广泛[9]。笔者在本文建立UPLC法,可同时检测罗汉果中的7种成分,为罗汉果药材及制剂的质量研究和控制提供可靠的分析方法。报道如下。
1 仪器与试药
1.1仪器 Agilent 1290超高效液相色谱仪(美国安捷伦公司);Agilent 6460 LC-MS/MS三重四极杆质谱仪(美国安捷伦公司);ME204E型电子天平(瑞士梅特勒-托利多公司,感量:0.1 mg);XP26型电子天平(瑞士梅特勒-托利多公司,感量:0.001 mg);KQ500D型数控超声波清洗器(昆山市超声仪器有限公司);TG-16高速离心机(四川蜀科仪器有限公司);Waters BEH C18色谱柱(50 mm×2.1 mm,1.7 μm)。
1.2药品与试剂 罗汉果皂苷V(含量:96.1%,中国食品药品检定研究院,批号:111754-202104);罗汉果对照药材(中国食品药品检定研究院,批号:121020-201807);罗汉果对照药材(北京万佳标准物质研究中心,批号:LG30025);11-O-罗汉果苷Ⅴ( 含量:98%,批号:PRF7102401),赛门苷Ⅰ(含量:98%,批号:PRF21012041),罗汉果苷Ⅳ(含量:98%,批号:PRF20082144),罗汉果苷Ⅲ(含量:98%,批号:PRF9013305),罗汉果苷Ⅲe(含量:98%,批号:PRF10050641),罗汉果苷Ⅱe(含量:98%,批号:PRF10083041),均购自成都普瑞法科技开发有限公司;乙腈为色谱纯,其他试剂为分析纯;罗汉果药材来源于多个不同产地,经华中科技大学同济医学院药学院尹春萍副教授鉴定为罗汉果原果。
2 方法与结果
2.1色谱与质谱条件 以Waters BEH C18(2.1 mm×50 mm,1.7 μm)为色谱柱;进样量为1 μL;柱温为35 ℃;紫外检测波长为203 nm;以水-乙腈(80:20)为流动相A,以水-乙腈(10:90)为流动相B, 梯度洗脱(0~2 min 100%A,2~10 min 100%A→90%A,10~11 min 90%A,11.01~16 min 100%B,16.01~18 min 100%A);流速为每分钟0.3 mL。质谱定性鉴别采用电喷雾离子化源(electrospray ionization,ESI);离子监测模式为选择离子监测(selected ion monitoring,SIM);监测的离子为1307.8,1309.8,1147.8,1147.8,985.7,985.7和823.7;干燥气温度 300 ℃;干燥气流速为每分钟10 L;雾化气压力为50 psi;鞘气温度为300 ℃; 鞘气流速为每分钟10 L;传输毛细管电压3500 V; 喷嘴电压1000 V。
2.2溶液的制备
2.2.1对照品溶液的制备 分别精密称取11-O-罗汉果苷Ⅴ、罗汉果皂苷Ⅴ、赛门苷Ⅰ、罗汉果苷Ⅳ、罗汉果苷Ⅲ、罗汉果苷Ⅲe、罗汉果苷Ⅱe对照品适量,用水-甲醇(40:10)溶解并稀释制成含罗汉果皂苷Ⅴ对照品浓度为50 μg·mL-1的溶液,其他对照品为5 μg·mL-1混合溶液,作为对照品溶液。
2.2.2供试品溶液的制备 将罗汉果原药材粉碎后,称取药材粉末约0.25 g,精密称定,置50 mL量瓶中,精密加入水40 mL,超声处理(功率500 W,频率40 kHz)40 min,取出,放冷,用甲醇定容,摇匀,取适量,13000 r·min-1(离心半径r=3 cm)离心5 min,取上清液,即得供试品溶液。
2.3方法学验证
2.3.1紫外光谱和质谱行为表征 称取各对照品适量,精密称定,用水-甲醇(40:10)溶解并定量稀释制成含各组分浓度约为0.1 mg·mL-1的混合对照品溶液,进样分析,采用二极管阵列检测器和质谱检测器进行检测,进行各组分的紫外吸收光谱(ultraviolet absorption spectra,UV)和质谱母离子扫描。采集各对照品主峰的UV光谱,各成分吸收峰波长在193 nm。为避免低波段较大的干扰,因此参考《中华人民共和国药典》2020年版(一部)罗汉果药材标准,选择波长203 nm为检测波长;母离子扫描质谱各组分均为[M+Na]+峰,质荷比分别为1307.8,1309.8,1147.8,1147.8,985.7,985.7和823.7,采用选择离子监测模式对上述离子进行检测。
2.3.2专属性实验 分别进样空白溶液(水-甲醇40:10)、混合对照品溶液和罗汉果药材溶液,记录色谱图,结果11-O-罗汉果苷Ⅴ、罗汉果皂苷Ⅴ、赛门苷Ⅰ、罗汉果苷Ⅳ、罗汉果苷Ⅲ、罗汉果苷Ⅲe、罗汉果苷Ⅱe依次出峰,空白溶剂对测定无干扰。色谱图见图1。
2.3.3检测限和定量限 精密称取对照品适量,用水-甲醇(40:10)配制各组分浓度约为0.5 μg·mL-1和0.25 μg·mL-1的混合对照品溶液,作为定量限(limit of quantitation,LOQ)和检测限(limit of detection,LOD)溶液,进样分析,计算各色谱峰的信噪比,定量限溶液色谱图中各色谱峰的信噪比均>10,检测限溶液色谱图中各色谱峰的信噪比均>3。
2.3.4线性关系考察 精密称取各对照品适量,用水-甲醇(40:10)配制成不同浓度的混合对照品溶液,在“2.1”项色谱条件下进样测定各组分的峰面积,以峰面积(A)为纵坐标,质量浓度(C)为横坐标进行回归,并计算各成分相对罗汉果苷Ⅴ的校正因子:校正因子=罗汉果苷Ⅴ线性斜率÷其他组分线性斜率。结果见表1。结果表明11-O-罗汉果苷Ⅴ、罗汉果皂苷Ⅴ、赛门苷Ⅰ、罗汉果苷Ⅳ、罗汉果苷Ⅲ、罗汉果苷Ⅲe和罗汉果苷Ⅱe在各自范围内线性关系良好。
2.3.5精密度实验 取对照品溶液,在“2.1”项色谱条件下,重复进样6次,记录色谱图,计算色谱图中11-O-罗汉果苷Ⅴ、罗汉果皂苷Ⅴ、赛门苷Ⅰ、罗汉果苷Ⅳ、罗汉果苷Ⅲ、罗汉果苷Ⅲe和罗汉果苷Ⅱe峰面积的RSD,结果分别为1.9%,1.6%,1.7%,0.6%,1.8%,1.5%和1.9%。
2.3.6稳定性实验 精密吸取罗汉果药材同一份供试品溶液,于0,13,23和24 h进样检测各组分的峰面积,计算峰面积的RSD,11-O-罗汉果苷Ⅴ、罗汉果皂苷Ⅴ、赛门苷Ⅰ、罗汉果苷Ⅳ、罗汉果苷Ⅲ、罗汉果苷Ⅲe和罗汉果苷Ⅱe峰面积的RSD分别为0.6%,1.3%,1.2%,1.1%,1.3%,1.2%和2.1%,供试品溶液在24 h内稳定。
2.3.7重复性实验 分别取同一批次罗汉果对照药材,按照“2.2.2”项下方法制备6份供试品溶液,依法进样测定11-O-罗汉果苷Ⅴ、罗汉果皂苷Ⅴ、赛门苷Ⅰ、罗汉果苷Ⅳ、罗汉果苷Ⅲ、罗汉果苷Ⅲe和罗汉果苷Ⅱe的峰面积,计算得7种成分含量的RSD分别为1.7%,1.3%,2.0%,2.3%,2.2%,2.2%,1.5%。
2.3.8回收率实验 称取罗汉果对照药材0.25 g,按照“2.2.2”项下制备供试品溶液并测定含量,作为原有量,平行测定3份,计算各组分原有量的平均值;另精密称取各对照品适量,用水稀释制成含罗汉果皂苷Ⅴ浓度为5 mg·mL-1的溶液(储备液A),11-O-罗汉果苷Ⅴ、赛门苷Ⅰ、罗汉果苷Ⅳ、罗汉果苷Ⅲ浓度为0.125 mg·mL-1的混合对照品溶液(储备液B),罗汉果苷Ⅲe和罗汉果苷Ⅱe浓度为0.125 mg·mL-1的混合对照品溶液(储备液C)作为回收率储备液。分别称取9份罗汉果对照药材0.25 g,每3份为一组,作为低、中、高浓度组,分别加入储备液A 0.25 ,0.5和1 mL(相当于罗汉果皂苷Ⅴ含量为0.5%,1.0%和2.0%),储备液B 0.2,2和8 mL(相当于11-O-罗汉果苷Ⅴ、赛门苷Ⅰ、罗汉果苷Ⅳ和罗汉果苷Ⅲ含量为0.01%,0.1%和0.4%),储备液C 0.2,1和2 mL(相当于罗汉果苷Ⅲe和罗汉果苷Ⅱe含量为0.01%,0.05%和0.1%),用水补足至40 mL,照“2.2.2”项下制备供试品溶液并按照各组分的对照品外标法进行测定,计算测得量,按下式计算回收率,回收率(%)=(测得量-原有量均值)/加入量×100%。分别计算平均回收率和RSD,结果见表2。另将回收率溶液色谱图中除罗汉果皂苷Ⅴ外其他组分的峰面积乘以按照表1中的校正因子后,用罗汉果皂苷Ⅴ对照品溶液按加校正因子的外标法测定各组分含量,计算平均值,结果对比见表3。
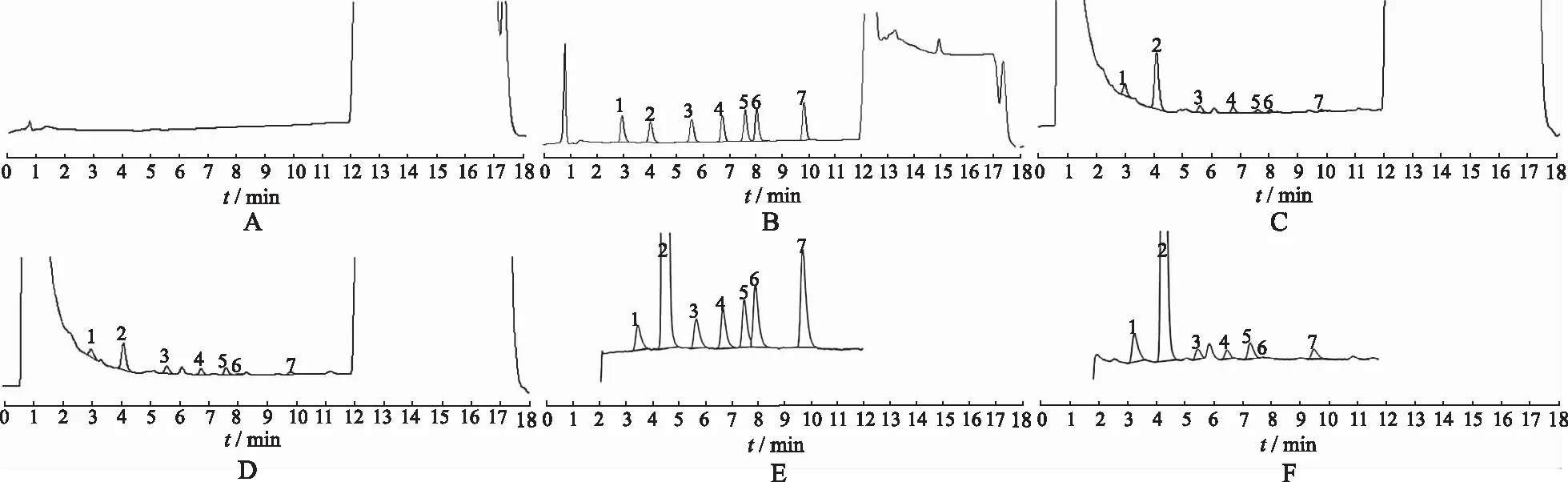
A.空白溶剂UV色谱图;B.混合对照UV色谱图;C.对照药材UV色谱图;D.罗汉果药材UV色谱图;E.混合对照SIM色谱图;F.对照药材SIM色谱图;1.11-O-罗汉果苷Ⅴ;2.罗汉果皂苷Ⅴ;3.赛门苷I;4.罗汉果苷Ⅳ;5.罗汉果苷Ⅲ;6.罗汉果苷Ⅲe;7.罗汉果苷Ⅱe。
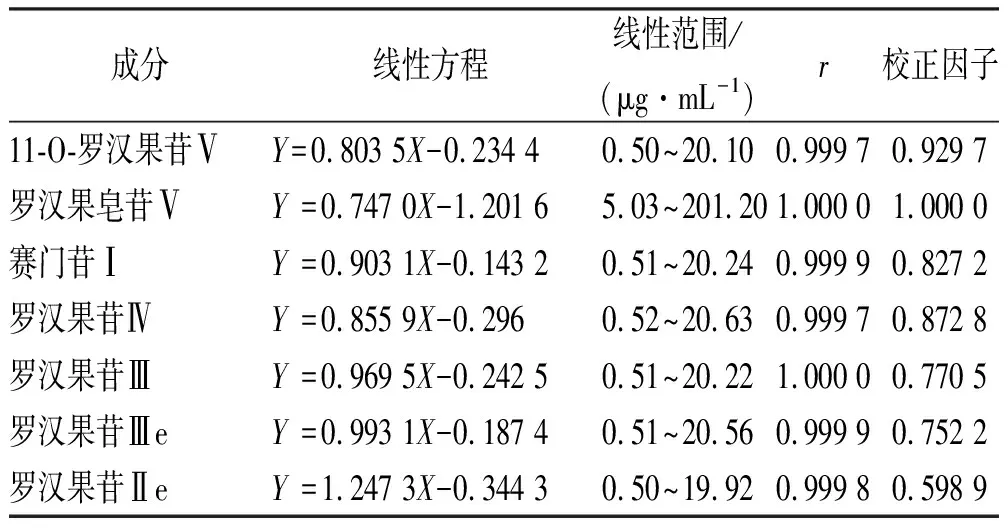
表1 7种成分线性关系与范围及校正因子
2.3.9相对保留时间的考察 以罗汉果皂苷Ⅴ峰为参照,分别考察不同条件下各组分的相对保留时间,包括调整流速为0.28和0.32 mL·min-1、柱温为33和37 ℃、A相为水-乙腈78:22(比例1)和82:18(比例2),结果见表4。
2.3.10样品测定结果 取26批罗汉果药材进行测定,计算各组分的含量,结果见表5。
3 讨论
3.1检测指标成分的选择 在《中华人民共和国药典》2020年版一部罗汉果药材标准中,采用罗汉果皂苷Ⅴ 作为罗汉果的含量指标,限度为不低于0.5%。该指标单一,无法全面反映罗汉果的药材质量。罗汉果中尚含有11-O-罗汉果苷Ⅴ,赛门苷Ⅰ、罗汉果苷Ⅳ、罗汉果苷Ⅲ、罗汉果苷Ⅲe及罗汉果苷Ⅱe等,因此,本文选择罗汉果皂苷Ⅴ等7种成分作为指标,进行罗汉果质量的考察。
3.2提取溶剂及提取方式的考察 《中华人民共和国药典》2020年版一部收载罗汉果含量测定方法,采用回流提取的方式进行提取。笔者分别考察水、甲醇、乙醇、异丙醇等不同溶剂,超声和回流提取等不同提取方式,以及不同溶剂体积的提取效果,结果表明以水为提取溶剂,采用超声提取的方式,即可有效提取出本品中的目标组分,相比《中华人民共和国药典》收载的提取方法简单易行。
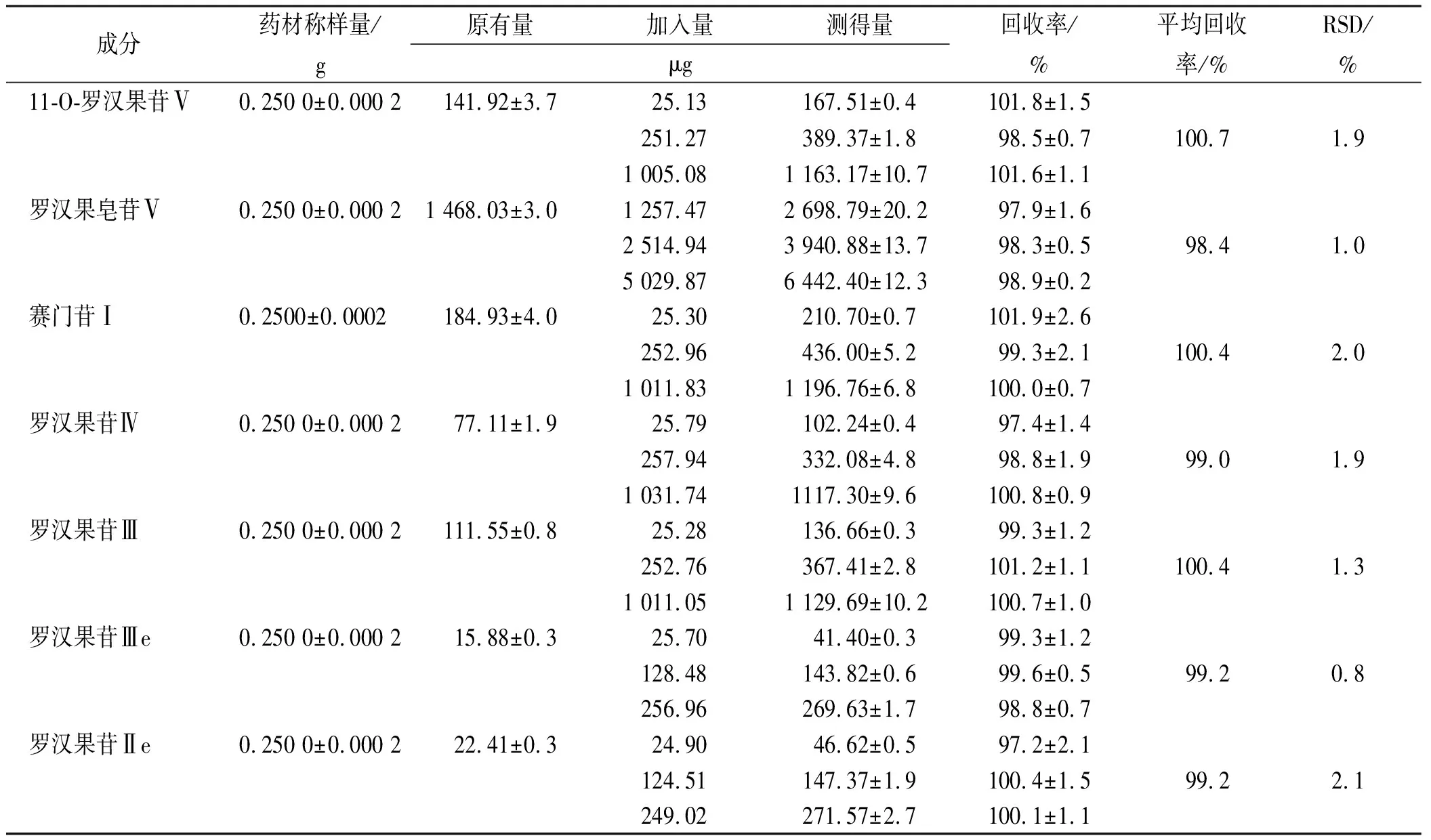
表2 7种成分回收率实验结果
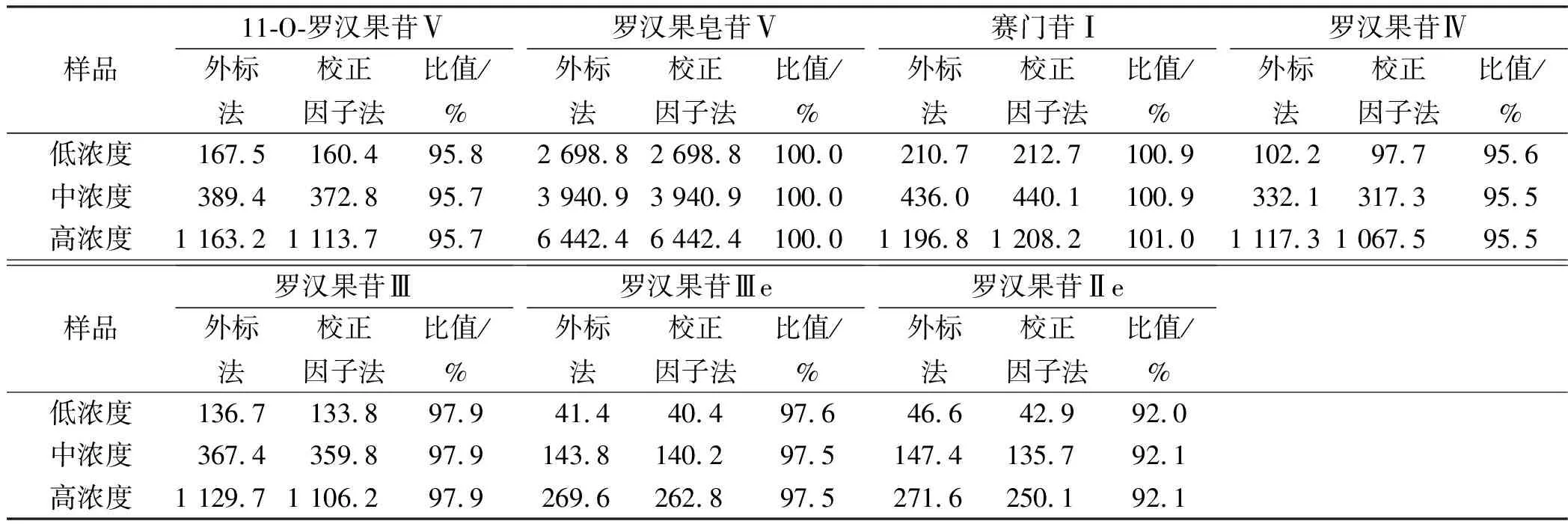
表3 两种定量方式的测定结果比较
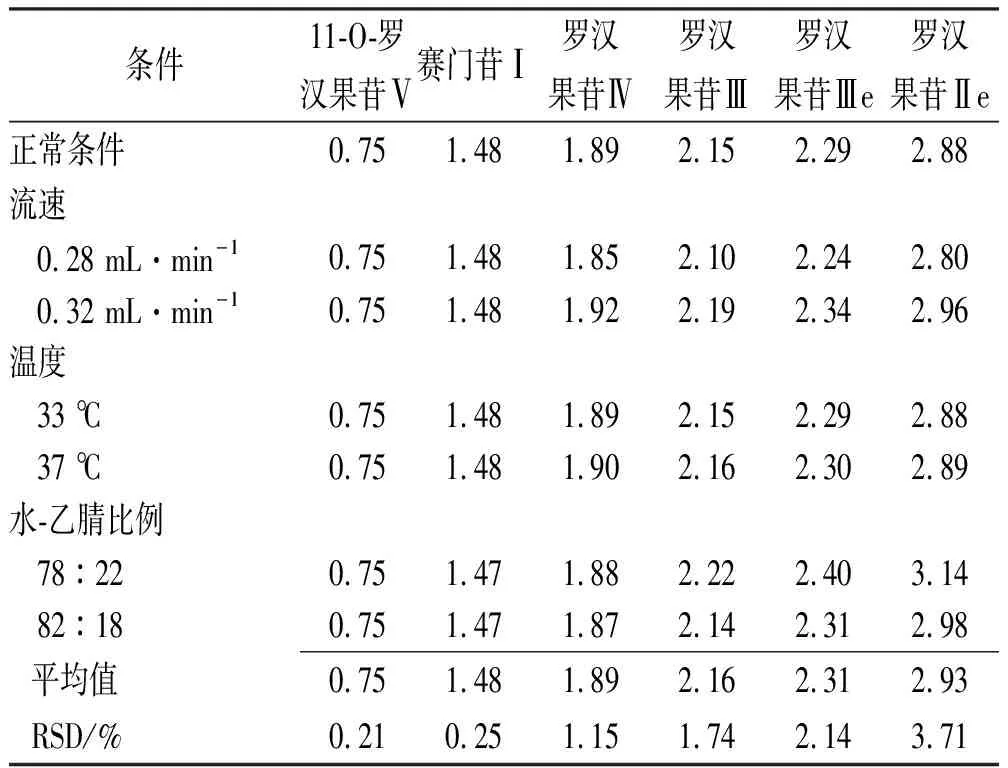
表4 6种成分相对保留时间的考察结果
3.3色谱条件的选择
3.3.1流动相的选择 由于各组分均为低波长吸收,因此采用截止波长较低的乙腈作为有机相;另外,为使流动相可兼容质谱分析,实现对目标组分进行准确定性,并为后续采用高分辨质谱对更多的其他未知组分进行结构鉴定预留空间,因此流动相中未添加不挥发性的缓冲盐。同时,由于甲酸、乙酸等挥发性缓冲盐在低波长下背景噪音较高,掩盖目标组分的色谱峰,因此也未使用。本文选用简单的乙腈-水体系作为流动相体系,各组分峰型良好。
在采用该流动相体系进行质谱分析时,出现信号很强的[M+Na]质谱信号峰,[M+H]信号峰极弱,且在进行碎片离子扫描时,碎片离子不明显,可能的原因是由于各组分无明显的酸碱基团,在ESI电离源下很难形成[M+H]或[M-H]信号,反而出现了较强的[M+Na]信号峰,而碎片离子也同样由于没有稳定的带电形式,质谱信号较弱。
3.3.2色谱柱和梯度的选择 本试验首先采用Agilent SB C18(4.6 mm×150 mm,3.5 μm)色谱柱,以高比例的水相作为起始比例,进行梯度洗脱,通过调整梯度变化速率以实现各组分的分离,但运行时间较长,因此选择柱效更好的超高效液相色谱柱,通过进一步优化梯度,建立了本方法。
3.3.3定量方式的选择 本文除采用对照品外标法进行含量测定外,尚测定各组分相对于罗汉果皂苷V的相对保留时间及校正因子,并对比采用对照品外标法和校正因子法测定结果的差异,结果表明两种计算方式的差异在10%以内,差异较小。因此,当实验室完成方法确认后,可按照相对保留时间对各组分进行定位,采用校正因子法对含量进行测定,节约对照品的使用。
3.4检测结果的分析 本文检测26种不同厂家和批次的罗汉果药材,其中罗汉果皂苷Ⅴ的含量最高,除批次R外,均能满足《中华人民共和国药典》2020年版一部收载的罗汉果标准的要求;11-O-罗汉果苷Ⅴ、赛门苷Ⅰ和罗汉果苷Ⅳ均在定量限0.01%以上,最小值为0.02%;罗汉果苷Ⅲ、罗汉果苷Ⅲe和罗汉果苷Ⅱe有部分样品低于0.01%,整体含量较低;11-O-罗汉果苷Ⅴ、罗汉果皂苷Ⅴ、赛门苷Ⅰ、罗汉果苷Ⅳ和罗汉果苷Ⅲ的测定结果与文献[8]的测定结果趋势基本一致,表明本方法测定结果准确可靠。基于26批次药材的检测结果,可以考虑将11-O-罗汉果苷Ⅴ、赛门苷Ⅰ和罗汉果苷Ⅳ增订入罗汉果药材的质量标准。另外,各组分的总量除K和R外,均>1%,也可酌情将总量订入质量标准。
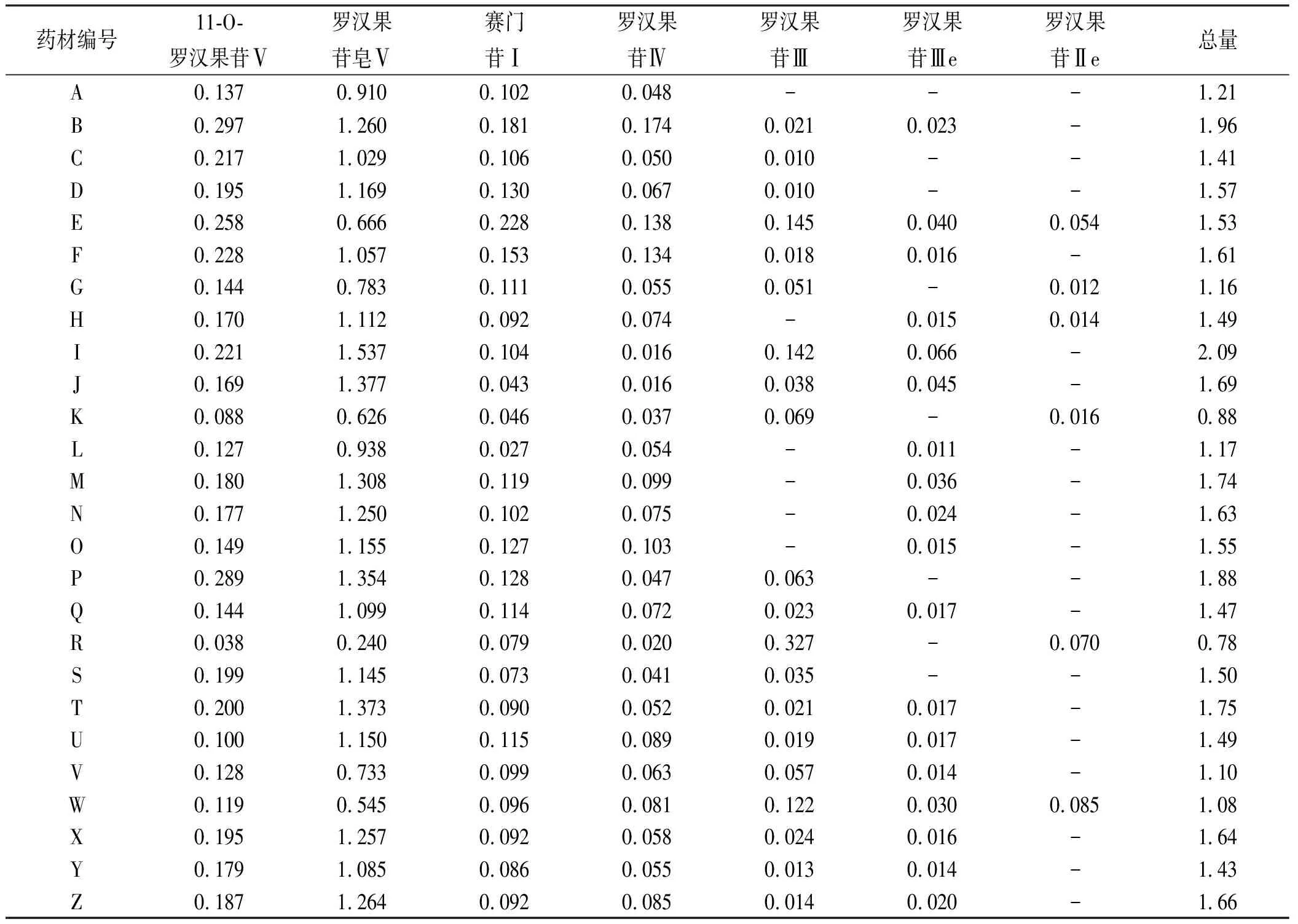
表5 26批罗汉果药材中7种成分的含量测定结果
笔者采用超高效液相色谱法,对罗汉果中7种组分进行测定,建立罗汉果中多组分的检测方法,有助于药品生产企业不断完善本品的质量标准,从药材的产地、种属、采收季节等方面全面研究罗汉果药材的质量情况,以达到有效成分含量最高,临床疗效最优的目标。本文建立的罗汉果中多组分的分析方法,时间短,专属性强,灵敏度高,为罗汉果药材的质量控制与标准提高提供了一定的指导意义。
