氮硫掺杂碳量子点荧光探针检测Fe3+和Hg2+
2022-10-08刘华东徐浩轩李合伟王定标
刘华东, 徐浩轩, 李合伟, 王定标
(郑州大学 机械与动力工程学院,河南 郑州 450001)
0 引言
随着工业发展,生活用水中的重金属离子对人类健康和环境造成严重威胁。Hg2+是最具毒性的重金属离子之一。Fe3+在人体中虽然含量少但发挥着重要作用。因此,对水溶液中Hg2+、Fe3+的测定很重要。传统的原子吸收法[1]、电化学法、原子发射光谱法等金属离子检测方法需要复杂的操作过程,而利用碳量子点(CDs)检测金属离子具有操作简单、灵敏度高等优点[2]。
碳量子点具有低毒性、良好的化学稳定性、环境友好和耐光漂白等特性。通过对CDs进行表面钝化和功能化不仅可以提高CDs的荧光量子产率(FLQY)还可以增加其表面官能团数量[3]。当使用S和N元素掺入碳点时,其荧光量子产率和性能都有明显提高。Liu等[4]以蛋氨酸和柠檬酸为原料,制备了N、S共掺杂的碳量子点,具有良好的稳定性和抗光漂白性能,由于其表面具有羟基、羧基、氨基而具有良好水溶性和抗强酸强碱能力。
近些年,因碳点具备的特性可用来检测各种金属离子和官能团[5],在传感和生物成像等领域的应用中表现出巨大优势。Ma等[6]使用红叶作为碳源,用水热法制备碳量子点可用于Fe3+的检测和细胞成像。Liu等[7]用草鱼鳞片为原材料,用微波法合成了硫掺杂的碳量子点,实现了对Hg2+的检测。
目前,多数碳量子点作为荧光探针都只能检测1种金属离子,而检测2种金属离子可以扩宽碳点在传感器方面的应用。本文以柠檬酸和硫脲为前驱体,通过水热法一步合成S、N共掺的碳量子点(N—S—CDs),用于Hg2+和Fe3+的检测。通过透射电镜(TEM)、傅里叶红外光谱(FTIR)和X射线光电子能谱(XPS)观察碳点形貌和分析表面官能团,检测其对Hg2+和Fe3+的选择性和灵敏度,并将N—S—CDs用于自来水和湖水中Hg2+、Fe3+的检测。
1 实验部分
1.1 材料与仪器
柠檬酸、硫脲、二氯甲烷、环己烷、二甲基甲酰胺(DMF)、无水乙醇、氯化钠、氢氧化钠、盐酸、浓硫酸(98%)购自天津永大公司。氯化钡、无水氯化钙、硫酸铜、氯化镁、三氯化铁、乙酸锌、乙酸锰、硝酸镉、硝酸铬、氯化亚铁、硝酸铅、硝酸汞购自麦克林公司。所用药品均为分析纯级。实验用水均为超纯水。
碳点在WGL-45B电热鼓风干燥箱中合成;合成碳点微观形貌通过FEI TalosF200s透射电子显微镜观察;傅里叶红外光谱由Nicolet 670傅里叶红外光谱仪(FT-IR)测得;荧光光谱和吸收光谱通过F-2700分光光度计和TU-1810紫外可见分光光度计分析;表面能谱通过K-Alpha Plus电子能谱仪分析。
1.2 N—S—CDs的制备
称取0.25 g柠檬酸和0.25 g硫脲溶解于20 mL超纯水中,超声处理30 min。将上述混合溶液转移至50 mL聚四氟乙烯反应釜,置于干燥箱,80 ℃加热8 h,自然冷却到室温。反应后原液在8 000 r/min的转速下离心10 min,收集离心上清液用0.22 μm滤膜过滤,然后用1 000 da的透析袋透析2次,透析后的溶液即为合成碳点溶液。合成碳点溶液在4 ℃下储存备用。
1.3 Hg2+和 Fe3+ 检测
将1 mL浓度为100 μmol/L各种金属离子溶液(Fe3+、Ca2+、Mg2+、Fe2+、Ni2+、Pb2+、Co2+、Zn2+、Cu2+、Cd2+、Cr3+、Hg2+)与1 mL稀释后的N—S—CDs溶液充分融合,静置20 min,然后用荧光分光光度计测量各溶液在激发波长为350 nm时的荧光强度。
将稀释后的N—S—CDs溶液中分别加入不同浓度的Fe3+溶液(0、5、10、30、60、90、100、120、180、210、270、330、400、500、600 μmol/L)和不同浓度的Hg2+溶液(0、5、10、30、60、90、100、120 μmol/L),测量混合溶液在350 nm激发光下的荧光强度。
1.4 荧光量子产率的计算
以硫酸奎宁(QY=0.54,0.1 μmol/L)为标准样品,计算碳点的QY[2]:
φs=φst(Ast/Ist)(Is/As)(ηs/ηst)2。
(1)
式中:φ为荧光量子产率;I和A分别为积分发射强度和光学密度;η为溶剂的折射率;下标s和st分别表示样品和标准样品。
2 结果分析
2.1 N—S—CDs的表征

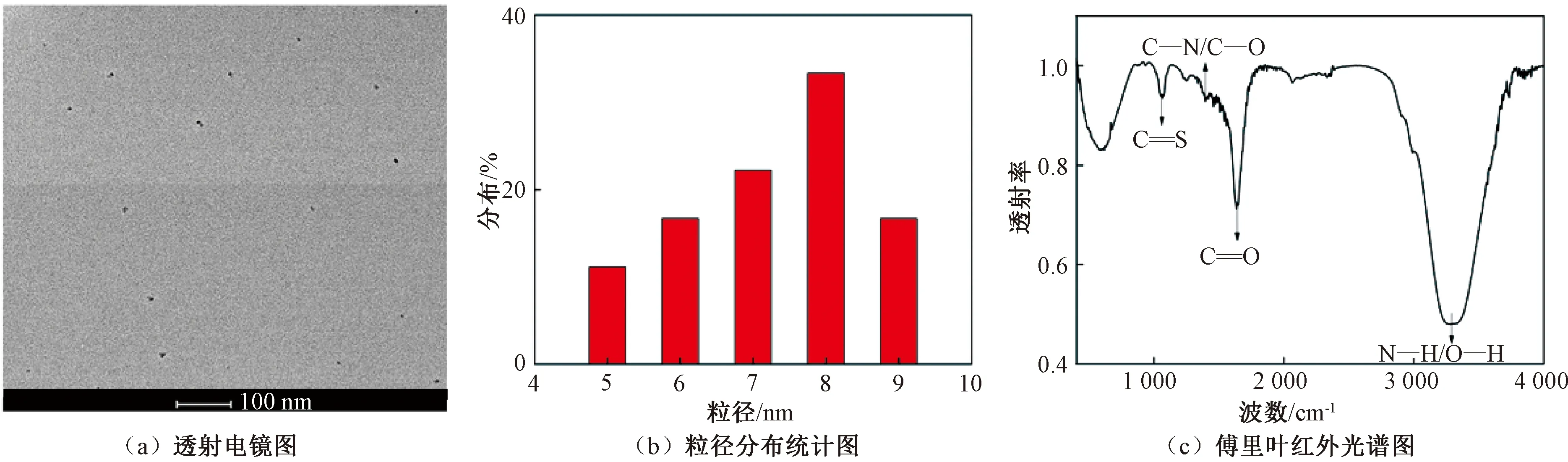
图1 N—S—CDs的透射电镜图及红外光谱图Figure 1 TEM and FTIR images of N—S—CDs
2.2 N—S—CDs的荧光性能
N—S—CDs溶液在紫外灯照射下发出很强的蓝色荧光,在自然光照下呈淡黄色。荧光光谱显示该N—S—CDs的最大激发波长为350 nm,荧光量子产率为36.8%,最大发射波长为440 nm(荧光强度最高),见图3(b);随激发波长从300 nm增大到390 nm,N—S—CDs的发射峰发生红移,呈现出先增强后减小的特征,在350 nm的激发波长下发射峰最强,见图3(c)。结果表明,合成碳点具有显著的激发波长依赖性,该依赖性是因为碳量子点表面发光位点不同或者不同粒径的碳量子点的尺寸效应[10]。
2.3 选择性研究
加入不同金属离子后N—S—CDs在350 nm激发光下的荧光强度见图4。从统计直方图中观察到,Hg2+和Fe3+对N—S—CDs有明显的荧光猝灭现象,其他金属离子猝灭程度很小。多数CDs发射荧光源于表面缺陷而捕获激子发生辐射复合,这些激子可以在CDs和金属离子之间进行非辐射电子转移而导致CDs荧光猝灭[11]。对于N—S—CDs,其表面含有大量官能团,与Hg2+或Fe3+配位形成络合物,引入更多缺陷作为激发能陷阱,导致电子从N—S—CDs向Hg2+或Fe3+转移发生荧光猝灭现象[7]。其中,Hg2+易与N—S—CDs中C—S结合,同时S原子的掺杂可以有效促进碳点和金属离子之间电子转移[12];Fe3+与N—S—CDs表面的—OH等含氧基团之间有良好的结合亲和力,促进碳点和金属离子之间的电子转移[13]。

图4 N—S—CDs选择性测试Figure 4 N—S—CDs selectivity test
当被吸附猝灭剂的吸收光谱与荧光碳点的激发和发射光谱有一定重叠时,会发生荧光共振能量转移(FRET)[14]。对比Hg2+和Fe3+的紫外-可见吸收光谱与N—S—CDs的激发、发射光谱,结果显示,Fe3+的吸收光谱与N—S—CDs的激发或发射光谱存在重叠,而Hg2+的吸收光谱与N—S—CDs的激发或发射光谱无重叠,见图5。
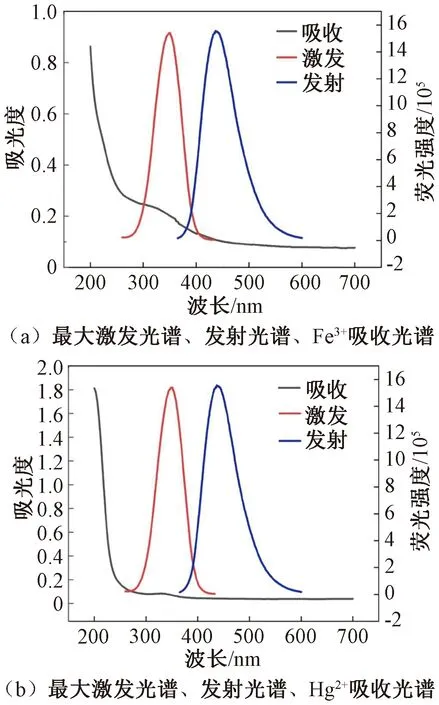
图5 N—S—CDs的荧光光谱与金属离子的吸收光谱Figure 5 Fluorescence spectra of N—S—CDs and absorption spectra of metal ions
2.4 Hg2+和 Fe3+灵敏度的检测
图6(a)是N—S—CDs荧光强度随Fe3+浓度增加的变化趋势图。当Fe3+浓度从0 μmol/L增加到600 μmol/L,荧光强度不断变弱,当Fe3+浓度达到180 μmol/L,猝灭程度达到了80%。当Fe3+浓度为40~130 μmol/L,N—S—CDs的荧光强度与Fe3+浓度呈线性关系,见图6(b),其线性拟合回归方程为F0/F=0.021 32cFe3++0.349 45,R2=0.995,F0和F分别为Fe3+加入前后的荧光强度。检出限使用公式 3Sd/k(Sd为21个平行测量的校正空白信号的标准偏差,k为校准曲线的斜率)[2]计算,估算Fe3+最低检出限为1.4 μmol/L。
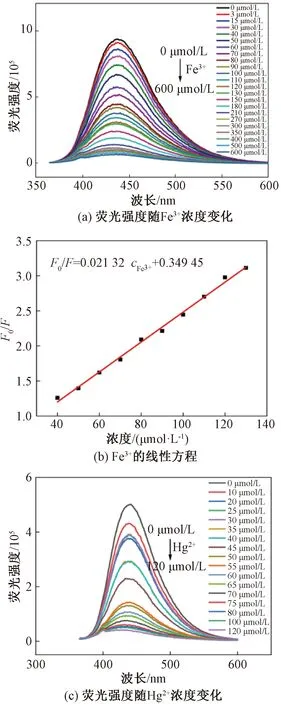

图6 N—S—CDs灵敏度测试Figure 6 N—S—CDs sensitivity test
随Hg2+浓度增加,N—S—CDs荧光强度变化趋势见图6(c),Hg2+浓度从0 增加到120 μmol/L,荧光强度不断变弱。其中,在40~80 μmol/L间,荧光强度与Hg2+浓度呈线性关系,线性拟合回归方程为F0/F=0.186 11cHg2+-6.271 14,R2=0.994,估算Hg2+的最低检出限为0.16 μmol/L。
2.5 实际水样检测
将N—S—CDs用于实际水样检测,所用自来水为郑州市自来水,湖水取自郑州大学眉湖。实际水样用0.22 μm注射过滤器进行过滤。用标准浓度的Hg2+和 Fe3+(45、50、55 μmol/L)进行回归实验(实验重复3次,取平均值),结果见表1。Fe3+的定量回收率为97.16%~103.62%,Hg2+的定量回收率为93.58%~101.22%。

表1 自来水和湖水中 Hg2+和Fe3+的回收率测试Table 1 Recovery tests for determination of Hg2+ and Fe3+ in tap water and lake water
3 结论
本文采用一步水热法,以柠檬酸和硫脲为前驱体,合成了一种氮、硫共掺杂的碳量子点(N—S—CDs)。N—S—CDs平均粒径为8 nm;在350 nm激发波长下发出蓝色荧光,荧光量子产率为36.8%;在中性和碱性的水溶液中能保持荧光稳定。可选择性检测Fe3+和Hg2+,线性方程为F0/F=0.021 32cFe3++0.349 45和F0/F=0.186 11cHg2+-6.271 14;灵敏度高,最低检出限分别为1.4 μmol/L和0.16 μmol/L。通过实际样品检测,N—S—CDs可以准确检测自来水和湖水中的Fe3+和Hg2+,Fe3+的定量回收率为97.16%~103.62%,Hg2+的定量回收率为93.58%~101.22%。
