Metabolic determinants of stemness in medulloblastoma
2022-10-08PaulaMartRubioPilarEspiauRomeraAlbaRoyoGarcLaiaCajaPatriciaSancho
Paula Martín-Rubio, Pilar Espiau-Romera, Alba Royo-García, Laia Caja, Patricia Sancho
Paula Martín-Rubio, Pilar Espiau-Romera, Alba Royo-García, Patricia Sancho, Hospital Universitario Miguel Servet, IIS Aragón, Zaragoza 50009, Spain
Laia Caja, Department of Medical Biochemistry and Microbiology, Biomedical Center, Uppsala University, Uppsala SE-751, Sweden
Abstract Medulloblastomas (MBs) are the most prevalent brain tumours in children. They are classified as grade IV, the highest in malignancy, with about 30% metastatic tumours at the time of diagnosis. Cancer stem cells (CSCs) are a small subset of tumour cells that can initiate and support tumour growth. In MB, CSCs contribute to tumour initiation, metastasis, and therapy resistance. Metabolic differences among the different MB groups have started to emerge. Sonic hedgehog tumours show enriched lipid and nucleic acid metabolism pathways, whereas Group 3 MBs upregulate glycolysis, gluconeogenesis, glutamine anabolism, and glutathione-mediated anti-oxidant pathways. Such differences impact the clinical behaviour of MB tumours and can be exploited therapeutically. In this review, we summarise the existing knowledge about metabolic rewiring in MB, with a particular focus on MB-CSCs. Finally, we highlight some of the emerging metabolism-based therapeutic strategies for MB.
Key Words: Medulloblastoma; Cancer stem cells; Stemness; Metabolism; Glycolysis;Lipids
lNTRODUCTlON
Medulloblastoma (MB) is the most common brain malignancy in young children. MB originates in the cerebellum and spreadsviathe cerebrospinal fluid to the brain and spine[1-3]. MB patients are classified in four different groups defined by the World Health Organization, based on the developmental pathway involved in their oncogenesis[2]: Wingless/integrated (WNT), sonic hedgehog (SHH), Group 3 and Group 4 (Table 1).
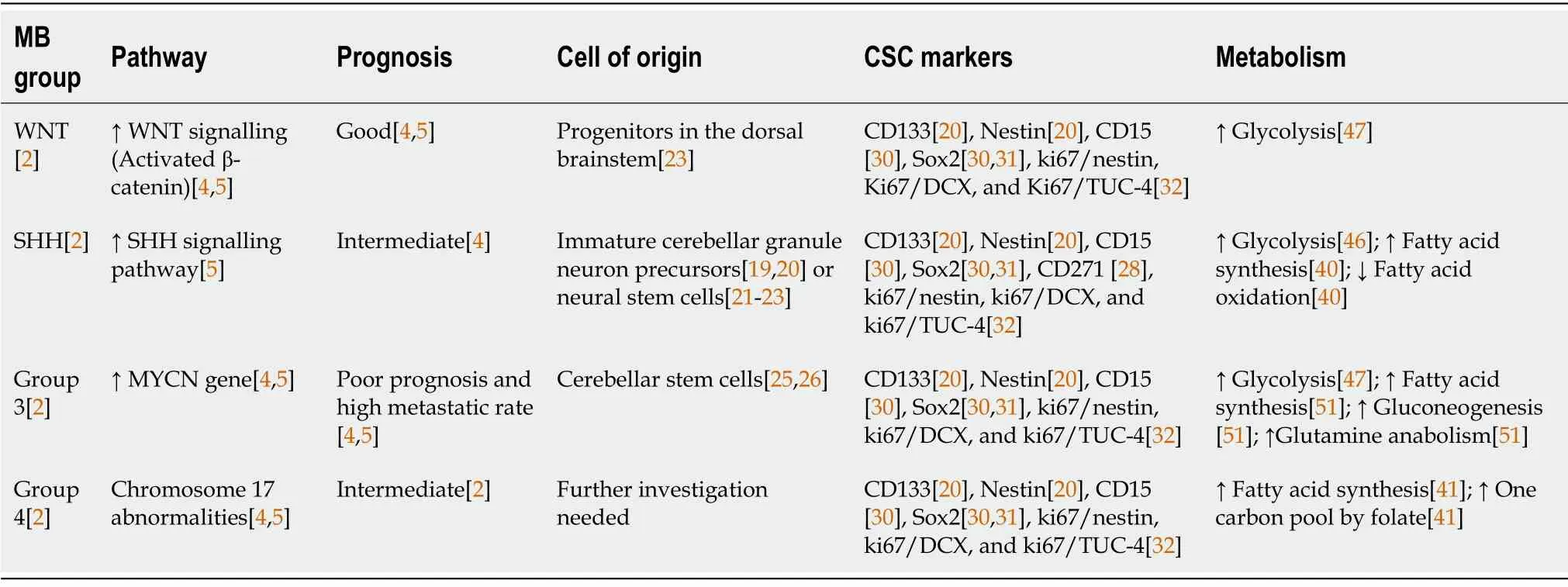
Table 1 Main features of medulloblastoma groups
The WNT group is the rarest, accounting for 10% of all MBs. These tumours occur in children over 3 years or teenagers, and generally have very good prognosis. WNT tumours show activating somatic mutations in β-catenin or germline mutations in adenomatous polyposis coli (APC), both leading to constitutive WNT signalling[4,5].
SHH group represents 30% of MB patients; most of them are either infants (3-years-old) or adults (>15-years-old), with intermediate prognosis[4]. The molecular mechanism regulating this subtype implicates loss-of-function mutations in negative regulators or over-expression of different members of the SHH signalling pathway such as protein patched homolog 1 (PTCH1), smoothened (SMO), GLI family zinc finger 1 (GLI), and suppressor of fused homolog (SUFU)[5].
Group 3 accounts for tumours with a poor prognosis and high metastatic rate, occurring predominantly in males (2:1) and young adults (> 16-years-old). These tumours display frequent amplification or overexpression of theMYCNgene, and other genetic events such as orthodenticle homeobox 2(OTX2) amplification, SWI/SNF related, matrix associated, actin dependent regulator of chromatin,subfamily A, member 4 (SMARCA4)mutation, andGFIenhancer activation[4,5].
Group 4 includes predominantly males (3:1), of all ages. These tumours show intermediate prognosis due to their metastatic potential: 30%-40% of Group 4 MB cases are already metastatic at diagnosis[2].No underlying common cause has been described for Group 4 tumours, but they show chromosome 17 abnormalities and neuronal differentiation transcriptomic profile[4,5].
CANCER STEM CELLS lN MB
The cancer stem cell concept
Cancers are hierarchically organised structures with different levels of intratumoural heterogeneity.Indeed, distinct clone and cancer cell populations co-exist in a tumour. Among them, there is a subset of cancer cells, the so-called cancer stem cells (CSCs), which are at the origin of intratumoural heterogeneity[6]. On the one hand, CSCs display the ability to self-renew, allowing them to preserve their identity as stem cells[6,7]. On the other hand, these cells maintain intraclonal heterogeneity and give rise to all differentiated progenies within each cancer subclone in the primary tumour[8]. These differentiated progenies have limited or no tumour-initiating and metastatic capacities, despite their high proliferation rate[9]. Additionally, CSCs have the enhanced capacity to drive tumorigenesis and progression,resistance to conventional radiotherapy and chemotherapy, and invasiveness[6,10]. In fact, they are primarily responsible for tumour metastasis in secondary organs[6]. Indeed, CSCs and tumour-differentiated cells can acquire mobility thanks to processes such as epithelial-to-mesenchymal transition. Both subpopulations can migrate to other tissues through the bloodstream, but only those with stem-like features will be able eventually to initiate secondary lesions causing metastasis[9].
Evidence of CSCs was first identified in acute myelogenous leukaemia[11] and was verified subsequently in other diseases such as breast cancer[12], pancreatic cancer[13], prostate cancer[14], as well as in brain tumours like MB[8]. Due to their malignant features, CSCs have become one of the main focuses for cancer therapy, with the aim of improving the understanding of cancer onset and progression, and finding new therapeutic strategies.
The origin of CSCs in the tumorigenesis process remains controversial, and it seems to be cancer typespecific. In general terms, two hypotheses have been formulated: The “stem cell hypothesis” and the“de-differentiation hypothesis”[7,15]. The “stem cell hypothesis” accounts for the similarities observed between normal tissue SCs and CSCs, postulating that malignant transformation driven by frequent genetic mutations in SCs[7,15] favours the aggressive behaviour of CSCs. The latter consists of a dedifferentiation of tumour cells caused by the acquisition of stem-like abilities and tumour-initiating potential[7,16].
MB-CSCs
Although the “de-differentiation hypothesis” cannot be completely ruled out, it is clear that the stem or progenitor populations represent the cell-of-origin of MB tumours and their comprised CSCs (Table 1).Indeed, the main genes and pathways altered in MB [WNT,SHH,Notch,MYC,signal transducer and activator of transcription 3 (STAT3)] are key regulators of cell cycle and stemness[17]. Early studies demonstrated that MB tumours originate from immature cerebellar granule neuron precursors[18,19] or neural SCs[8,20,21] escaping self-renewal restriction upon activation of the SHH pathway. While this still holds true for the SHH subgroup, further studies have determined that the different subgroups arise from distinct developmental origins. Indeed, the WNT group originates from progenitors in the dorsal brainstem[22] and Group 3 tumours derive from cerebellar SCs[23,24]. However, the origin of Group 4 MB is still under debate.
The original identification of brain CSCs in MB and other tumours involved the expression of the neural stem cell markers cluster of differentiation 133 (CD133) and Nestin[20] (Figure 1), enriching for a subpopulation of cells with enhanced neurosphere formation ability. These results were further developed in a subsequent study demonstrating that injecting as little as 100 CD133+MB cells produced a tumour resembling the patient’s original tumour[8], definitively linking CD133 expression with CSCs in MB. In further studies, CD133+MB cells have been characterised by their radiation[25] and chemotherapy[26] resistance, as well as their enhanced invasiveness[27,28]. Importantly, CD133 expression predicts poor prognosis in MB patients[29], associated with higher rates of recurrence and metastasis[26], especially in Group 3.
Besides the well-established role of CD133, the surface marker cluster of differentiation 15 (CD15,SSEA-1) was found as a marker of MB-CSCs in two independent studies using thePatchedmutant mouse model of SHH MB (Figure 1)[30]. In both studies, CD15+cells showed high tumorigenicity in transplantation experiments, giving rise to tumours histologically similar to the original ones. However,these studies contrasted in the definition of CD15+cells as progenitor or stem cell-like tumour cells,considering their CD133 expression and their ability to form neurospheres composed by different cellular subpopulations. Interestingly, SRY-box transcription factor 2-positive (Sox2+) cells form a quiescent subpopulation within CD15+cells with enhanced tumour-initiating abilities[31] and both Sox2+and CD15+signatures were associated with poor prognosis and lower survival rates in patients[30,31]. Noteworthy, CD15+signature was not restricted to any specific MB subgroup, suggesting that this marker predicts poor survival in MB patients, in general[30].
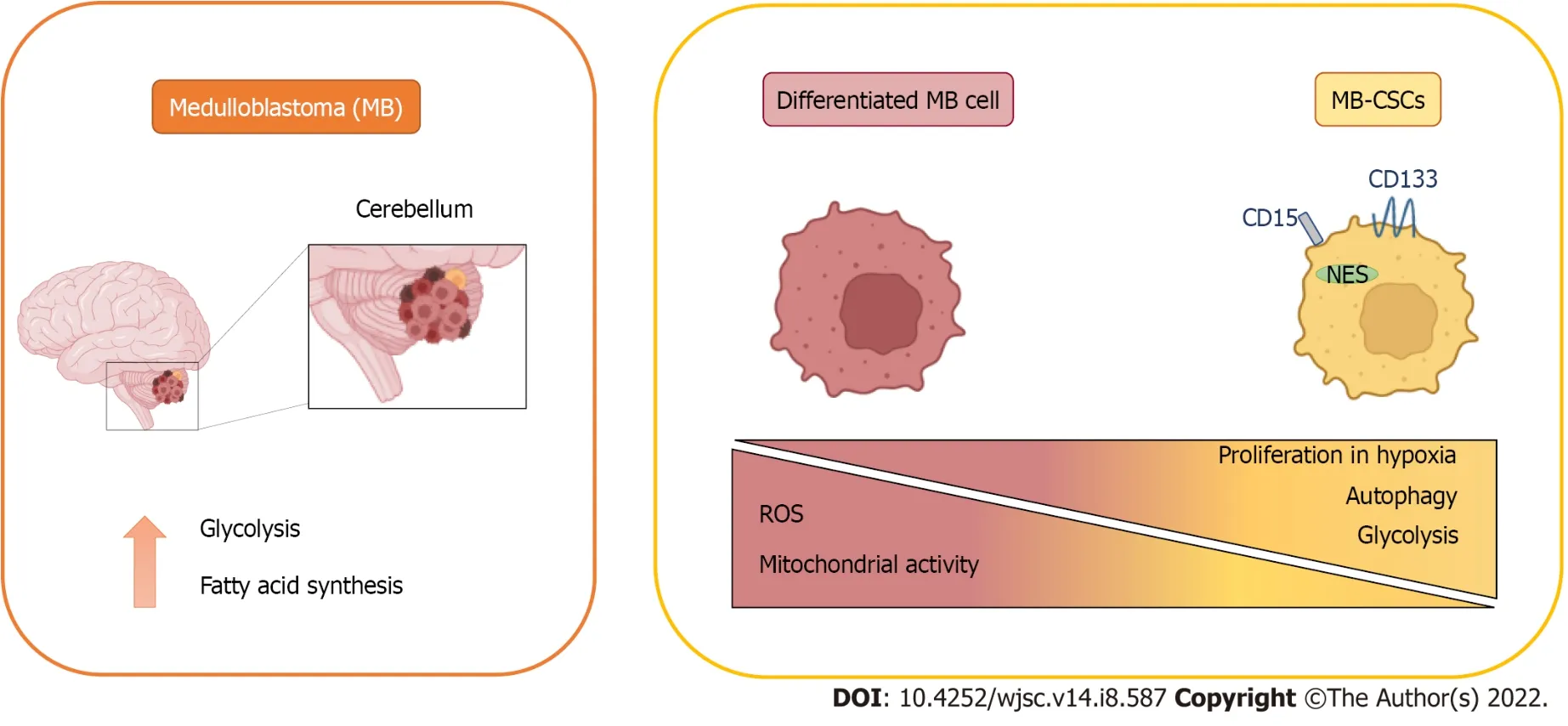
Figure 1 Metabolic features of medulloblastoma and medulloblastoma cancer stem cells.
Interestingly, the specific expression pattern of CSC markers can distinguish the functional heterogeneity of MB-CSCs in less aggressive subtypes such as SHH[28]. Indeed, isolation of diverse clones from the well-studied SHH cell line DAOY revealed that highly self-renewing cells expressed CD271[p75 neurotrophin receptor (p75NTR)], while those with enhanced migratory capacity expressed CD133.However, CD271 expression is much lower in aggressive tumours from Groups 3 and 4, and does not allow for functional classification of their MB-CSCs.
Highly proliferative ki67+CSCs have been also linked to worse prognosis, recurrence, and metastasis in MB patients[32]. Although the percentage of cells expressing CD133 was increased only slightly in relapsed tumours, authors found that the most significantly altered parameter in recurring tumours was the proliferative index of CSCs, calculated as the percentage of ki67+CSCs, especially in distant metastases.
Additionally, cells with CSC-like properties have been identified in MB patient samples using the side population (SP) technique, which entails the evaluation of the Hoechst 33342 dye exclusion by flow cytometry. Isolated MB-SP cells showed an increased neurosphere formation ability, as well as an overexpression of the stem cell markers Nestin, Notch1, and ABCG2[33].
METABOLlC FEATURES OF MB CANCER (STEM) CELLS
MB tumour cells must be provided not only with the essential metabolites to cover their energy requirements, but also with the macromolecules demanded for tumour growth. Consistent with this,increased lipogenesis and aerobic glycolysis have been detected in MB[34]. However, MB metabolic profile is not uniform among the different subtypes (Figure 1)[35].
Lipid metabolism in MB
SHH oncogenic signalling inhibits fatty acid oxidation while increasing fatty acid synthesis, an early critical step of lipogenesis, to promote tumour grow. Indeed, extensive lipid accumulation and elevated levels of the lipogenic enzyme fatty acid synthase (FASN) were detected in SHH-driven MBs from transgenic mice. In fact, the Rb/E2 factor (E2F) tumour suppressor complex acted downstream of SHH,controllingFASNexpression, and promoting lipogenesis and tumour growth[36].Furthermore,FASNand stearoyl-CoA desaturase (SCD), both with crucial roles in the fatty acid synthesis pathway, were identified as prognostic genes in the SHH subgroup, Group 3, and Group 4 of MB (Figure 2)[37].
Glucose metabolism in MB
In many cancers, energy metabolism shifts from oxidative phosphorylation to aerobic glycolysis for ATP production, phenomenon known as the Warburg effect[38]. Several studies indicate that this effect also occurs in MB[39-41].
High expression levels of the oncogenec-MYC, which is a downstream target of the WNT signalling pathway and is overexpressed in Group 3 MB, has been related with increased glycolysis in MB[41-43].Indeed, MBs from Groups 3 and WNT express significantly higher levels of the glycolytic enzyme lactate dehydrogenase A (LDHA) downstreamc-MYCthan non-neoplastic cerebellum (Figure 2).Interestingly,LDHAlevels have been linked with poor prognosis in Group 3 MBs[43]. BesidesLDHA,other glycolytic genes, such as hexokinase 2 (HK2) and pyruvate kinase M2 (PKM2), are upregulated both in murine MYC-amplified and human Group 3 MBs.PKM2upregulation is also associated with a subsequent downregulation of thePKM1isoform, which promotes oxidative phosphorylation in MYCamplified MB tumour cells[42].
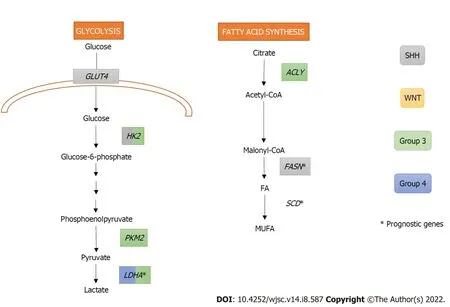
Figure 2 Metabolic pathways upregulated in medulloblastoma.
Induction of glycolysis in SHH MBs requires activating both SHH and insulin/insulin growth factor(IGF)/phosphoinositide 3-kinase (PI3K) pathways[40]. In addition, downstream activation of Myc-Max effector complex is also necessary forHK2upregulation. Interestingly, rather thanLDHAupregulation,SHH subgroup showed higher levels ofLDHBthan the other groups[41] suggesting thatLDHBcould be performing the expected role ofLDHA[40]. Peroxisome proliferator-activated receptor gamma (PPARγ),a key regulator of fatty acid and glucose metabolism, is also upregulated in SHH-induced mouse MBs.This transcription factor, controlled by E2F1, induces the expression of key glycolytic enzymes includingHK2,PKM2, and glucose transporter type 4 (GLUT4)[39].
Although increased uptake of18F-fluoro-2-deoxyglucose, a readout of aerobic glycolysis, has been detected in human MB tumours[44] and tested in clinical trials for MB detection and follow-up (i.e.NCT00381797), some evidence questions the universality of the Warburg phenotype in MB. On the one hand, an increased expression of genes associated with the mitochondrial respiratory chain and oxidative phosphorylation was detected in human SHH MBs, but not in mouse SHH-MB models. This suggests that murine SHH-MB models do not recapitulate the metabolic features of human MBs[45]faithfully. On the other hand, a recent publication also questions the Warburg effect in MYC-amplified MBs: higher glucose incorporation into the tricarboxylic acid (TCA) cycle was detected in orthotopic tumours compared with normal brain, indicating that tumour cells use glycolysis and oxidative phosphorylation simultaneously. In addition, significant differences in glucose and glutamine metabolism were detected in high MYC-amplified MBs comparingin vitro, flank xenografts, and orthotopic xenograft MB models, evidencing the importance of using orthotopic models for metabolic analysis in these tumours[46].
Other metabolic pathways altered in MB
Group 3 MB tumours not only display an enrichment of the glycolytic pathway but also the upregulation of proteins involved in other metabolic pathways such as gluconeogenesis (PCK2, PC), glutamine anabolism (GLUL), calcium signalling (RYR3, CAMK2D), fatty acid synthesis (ACLY), glutathionemediated antioxidant pathway (PTGDS, GSTZ1), and the drug metabolism pathway (IMPDH1, GUSB)[47]. In addition, the upregulation of the TCA cycle, synthesis of nucleotides, hexosamines, amino acids,and glutathione was also detected in MYC-amplified orthotopic xenograft MBs[46]. This highly metabolic profile could be responsible for the aggressiveness and chemoresistance of Group 3 and MYCamplified tumours.
Importantly, several metabolic pathways with prognostic value have been identified in MB including alanine, aspartate, and glutamate metabolism in SHH subgroup, pentose phosphate pathway and TCA cycle in Group 3, and one carbon pool by folate in Group 4[37].
Metabolism in MB-CSCs
Thanks to recent advances, we now know that cellular metabolism and stemness are highly interconnected in normal development and cancer. Indeed, CSCs from different cancer types show distinct metabolic features when compared with their more differentiated progenies, though their dominant metabolic phenotype varies across tumour entities, patients and even subclones within a tumour[48]. In fact, it has been shown that, depending on the type of cancer, CSCs may be either highly glycolytic or oxidative phosphorylation-dependent[49]. Moreover, recent evidence from both pluripotent embryonic and adult stem cell studies suggests that reactive oxygen species (ROS) in synchrony with metabolism mediate the maintenance of CSC[50]. A major role for the metabolic regulator mTOR in stemness maintenance has also been described in MB-CSCs[51,52]. Surprisingly, although MB stem cells are thought to be responsible for chemoresistance and recurrence in this cancer[53], their metabolic properties have barely been studied.
A decreased oxidative phosphorylation, mitochondrial mass and endogenous ROS production have been detected in established radioresistant MB stem-like clones (rMSLCs). These clones showed a higher pyruvate kinase activity and lactate production than parental cells. Mass spectrometry also detected an increased concentration of glycolysis intermediates (3-phosphoglyceric acid, 2-phosphoglyceric acid,phosphoenolpyruvic acid) and amino acids (arginine, glycine, histidine, leucine, lysine, serine) in rMSLCs, as well as decreased levels of NADH and ATP, and dysregulation of TCA cycle intermediates.Additionally, they showed an increased rate of conversion of pyruvic acid into lactic acid, and a lower rate of conversion of pyruvic acid into acetyl-CoA, which indicated an increased glycolysis[54].
MB cells expressing high levels of BMI1 (a key regulator of neural stem cells) and low chromodomain-helicase-DNA-binding protein 7 (CHD7) (an ATP-dependent chromatin remodeller)showed a decreased mitochondrial function concomitant with higher glycolytic activity. Moreover,enhanced expression of glycolytic genes [HK2,PFKP, enolase 4 (ENO4), pyruvate dehydrogenase kinase 1(PDK1) andLDHB] and increased levels of valine and leucine were detected in BMI1highCHD7lowGroup 3 MB tissues. Since both markers have been previously related to hypoxia, it was proposed that cells with this signature might present advantages in hypoxic conditions thanks to these metabolic adaptations (Figure 1)[55].
Indeed, oxygen restriction in non-vascularised regions may support cell cycle entry and self-renewalviaNotch1 signalling, since hypoxia is one of the main expansion inducers of the CD133+population[25,56]. In contrast, MB-CSCs associated to a more quiescent state have been reported to reside in a perivascular niche[57], in close interaction with endothelial cells that secrete factors to maintain their stemness.
Finally, it has been demonstrated that autophagy positively regulates stemness in MB cells. Indeed,the expression of the pro-autophagy factor AMBRA1 depends on c-MYC levels, and it is strongly upregulated in Group 3 MB-CSCs[58].
In general, all these reports suggest that the metabolic phenotype harboured by MB-CSCs is mainly glycolytic, which is favoured by hypoxia. Both glycolysis and hypoxia generate the antioxidant redox status needed to maintain their stemness. Moreover, the elevated availability of macromolecules needed to support their enhanced proliferation may be sustained by biosynthesis derived from glycolysis intermediates and an increased autophagy in these cells.
TARGETED THERAPlES lN MB
Standard therapies for MB
Current standard treatments for MB include surgical resection of the tumour, craniospinal radiotherapy and systemic chemotherapy. Unfortunately, only 70% of patients survive. In addition, radiotherapy causes neurocognitive, neuroendocrine and psychosocial deficits, as well as the development of secondary malignancies[59,60]. Consequently, new treatment strategies for MB are needed, such as targeted therapies that are less toxic and more effective in reducing the resulting long-term side effects[61].
Molecular targeted therapies for MB
Targeted therapies for WNT and SHH MB entail inhibiting the major pathways implicated in their tumourigenesis[62]. As an example, the use ofSmoinhibitors in the SHH MB group has been linked to tumour suppression in mouse models, and tumour regression in MB patients[62,63]. On the other hand,research on WNT MBs has focused on nuclear β-catenin inhibitors, leading to reduced cell migration and invasion[63,64].
In Groups 3 and 4 there are no major advances in targeted therapy as they have a low somatic mutation rate[61,63]. Group 3 MB is characterised by MYC oncogene amplification, so some epigenetic MYC inhibitors, such as histone deacetylases (HDACs) and bromodomain inhibitors, have been tested[65,66]. In addition, PI3K inhibitors can act in synergy with HDAC modulators, preventing tumour proliferation and prolonging survival in Group 3 MB mice[67].
Apart from subgroup-specific therapies, other molecular approaches linked to the general population of MB patients are being explored nowadays. Selective inhibition of cell division protein kinase (CDK)4/6 arrests the cell cycle potently and is therefore a favourable target for cancer treatment[68]. Another strategy is related to immunotherapy, as it may be effective against MB innate immunosuppressive properties and thus it may extend patient survival[69].
Metabolic targeted therapies for MB
Thoroughly investigated in the context of other cancer types, metabolic reprogramming might be also the source of putative targets for the design of new treatments against MB (Table 2). For example,specifically in lipid metabolism, direct inhibition of FASN by C75 reduced tumour proliferation in SHH tumours and increased survivalin vivo, associated to reduced lipogenesis. In addition, C75 may act in synergy with the CDK inhibitor roscovitine[36].
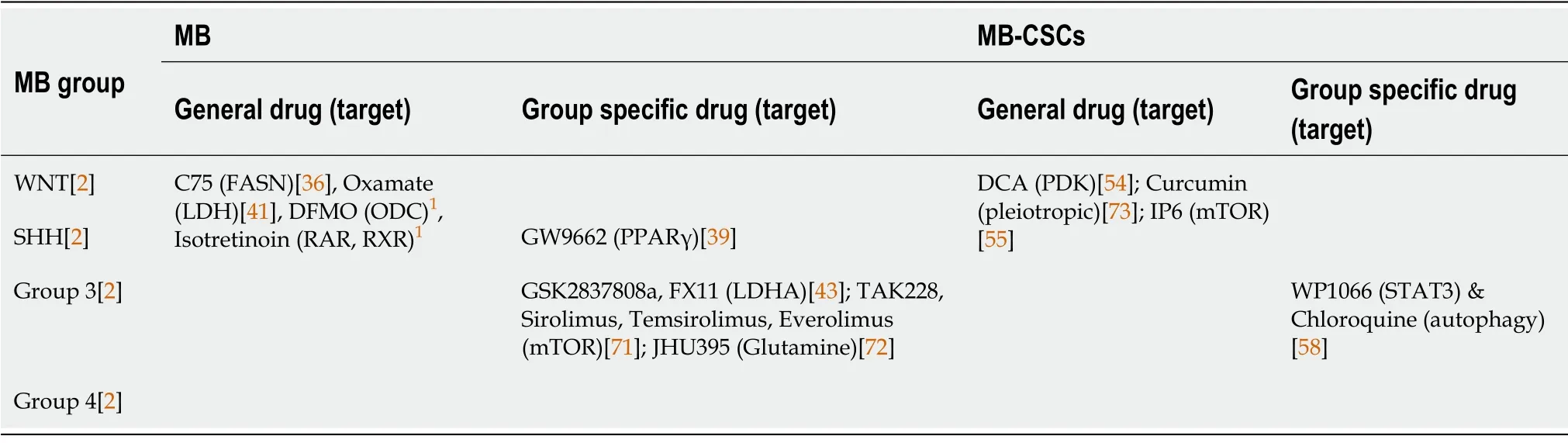
Table 2 Metabolic therapeutic targeting in medulloblastoma
Since glycolysis seems crucial for the metabolic requirements of MB, it may be important to target glycolytic enzymes. In that regard, LDH tetramers inhibition by oxamate attenuated glycolysis, proliferation, and motility significantly in MB cell lines[41]. In addition, LDHA antagonists GSK2837808a and FX11 reduced the growth of Group 3 MB cells[43]. Conditional suppression of HK2 altered both energy homeostasis and the balance between proliferation and differentiation markedly, thereby reducing tumour growth. Similarly, blockade of IL6 by bazedoxifene showed glycolysis downregulation, and impaired viability and proliferation of MB cells[70]. Finally, it is important to mention that pharmacological inhibition of PPARγ by GW9662 increases SHH-driven MB cell death and mouse survivalin vivothrough glycolysis inhibition[39].
Another interesting therapeutic strategy for MB is based on the inhibition of the metabolic regulator mTOR. Indeed, the TORC1/2 kinase inhibitor TAK228 improved the survival of orthotopic MYCamplified Group 3 MB tumours through glutathione depletion[71]. In addition, the dual inhibition of PI3Kα and mTOR blocked the sphere-forming ability of SHH-driven MB cells, and repressed tumour growthin vivo[52]. In fact, several clinical trials are currently testing different conventional treatments in combination with mTOR inhibitors, such as Sirolimus (NCT02574728), Temsirolimus (NCT00784914) or Everolimus (NCT03387020) against MB and other brain tumours.
The elevated MYC expression characteristic of Group 3 MB is related to an increased glutamine transport and utilisation. For this reason, the glutamine antagonist 6-diazo-5-oxo-l-norleucine has been tested as therapy against Group 3 MB, although with little success in clinical trials due to its poor pharmacodynamic properties. Still, the prodrug isopropyl 6-diazo-5-oxo-2-[phenyl (pivoxyloxy)methoxy-carbonyl] hexanoate (JHU395) has shown an improved cell penetration, enhanced growth reduction and increased apoptosis of Group 3 MB cellsin vitro, as well as longer survival of micein vivo[72].
Different metabolic targeting approaches related to the toxic effect of ROS accumulation and redox imbalance have also been tested in clinical trials. On the one side, two clinical trials have studied the antitumoral properties of Photodynamic Therapy, which uses light and photosensitizing drugs to kill tumour cellsviaROS accumulation, in MB (NCT00002647, NCT01682746). Unfortunately, no results have been posted to date. On the other hand, the antiprotozoal compound Nifurtimox, with pro-oxidant properties, has also been approved by the Food and Drug Administration for its use in MB. In the Phase II trial NCT00601003, the combination of Nifurtimox with cyclophosphamide and topotecan is being tested as treatment for relapsed or refractory neuroblastoma and MB.
There are other alternative therapeutic approaches targeting MB metabolism currently being tested in clinical trials. On the one hand, the polyamine synthesis inhibitor difluoromethylornithine (DFMO) is being tested in a phase II clinical trial as maintenance therapy for high molecular risk or very high risk and relapsed or refractory MB (NCT04696029). On the other hand, the combination of Vorinostat and Isotretinoin (13-cis-retinoic acid, vitamin A derivative) has been tested in a phase I clinical trial targeting young patients with recurrent or refractory lymphoma, leukemia, or solid tumours such as MB(NCT00217412), although no results have been posted to this day.
Targeted therapies for MB-CSCs
Though much less studied in the context of MB, metabolic pathways also play a key role in cancer resistance linked to CSCs. Treatment with the PDK inhibitor DCA suppressed CSC-like phenotypes, and increased intracellular ROS levels and radiosensitivity by inhibiting glycolysis and inducing mitochondrial aberrations[54]. In addition, the polyphenolic compound Curcumin, known to exert antitumor effects in many different cancer types, decreased anchorage-independent clonogenic growth and CD133+stem-like population by inhibiting STAT3 and PI3K/AKT pathways[73]. Furthermore,direct inhibition of the PI3K/AKT pathway with Perifosine resensitised MB-CSCs to radiotherapy by reducing CD133+MB stem-like subpopulations[74]. Inositol hexakisphosphate (IP6) treatment counteracted metabolic adaptation in BMI1HighMB cells thus inhibiting their proliferation. Moreover, IP6 synergised with Cisplatin, potentiating its cytotoxic activityin vitroandin vivo,and extending survival of the mice[55] significantly. Finally, pharmacological inhibition of the pro-autophagy factor AMBRA1 affected both the growth and invasion potential of group 3 MB-CSCs profoundly, enhanced by the combination of AMBRA1 and STAT3 inhibition[58].
Besides metabolism, we can also find other targets, such as the new SHH inhibitor Chemotype 12 that overcomes the associated SMO receptor drug resistance. It reduced the ability to form spheres in number and size, associated with a reduced expression of stemness markers [NANOG, octamer-binding transcription factor 4 (OCT4)], and thus impaired stem-like cell growthin vitroandin vivo[75].Moreover, CDK inhibitors, such as Ribociclib, can also target CSCs[68]. Oncolytic engineered herpes simplex viruses (oHSVs) may also have the potential to eliminate MB-CSCs specifically, as they express the primary HSV-1 entry molecule nectin-1 (CD111) and are shown to be highly sensitive to clinically relevant oHSVsin vitroandin vivo[76].
CONCLUSlON
Further research is urgently needed to elucidate the metabolic profile of MB tumours. Recent evidence suggests that metabolic features among MB groups and even among the different cell subpopulations within a tumour are highly heterogeneous. In addition, new data obtained in more clinically relevant models challenge most of the existing knowledge in the field, obtained from genetically modified mouse models. Moreover, the metabolic features of MB-CSCs remain absolutely unknown nowadays. The identification of metabolic vulnerabilities of these tumours and their highly aggressive CSCs could pave the way to the design of new therapeutic strategies improving the current treatment for this paediatric disease.
ACKNOWLEDGEMENTS
We want to thank Laura Sancho for proofreading the manuscript.
FOOTNOTES
Author contributions:Martín-Rubio P, Espiau-Romera P, Royo García A, Caja L, and Sancho P drafted the manuscript; Caja L and Sancho P designed the study and wrote the final version of the manuscript; Martín-Rubio P designed the figures; All authors approved the final version of the manuscript.
Supported bythe Miguel Servet and pFIS fellowships, No. CP16/00121 (P.S.) and No. FI21/00031 (P.E-R.) from the Instituto de Salud Carlos III and cofinanced by European funds (FSE: “el FSE invierte en tu futuro”); Magnus Bergvalls Stiftelse, No. 2021-04284 (L.C.); and the IV Grant for Childhood Cancer Research from Asociación de Padres de Niños con Cáncer de Aragón (ASPANOA, P.S.).
Conflict-of-interest statement:There are no conflicts of interest to report.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:Spain
ORClD number:Paula Martín-Rubio 0000-0002-2702-9212; Pilar Espiau-Romera 0000-0002-7165-199X; Alba Royo-Garcia 0000-0002-5079-6890; Laia Caja 0000-0002-8786-8763; Patricia Sancho 0000-0002-1092-5395.
S-Editor:Chen YL
L-Editor:Filipodia
P-Editor:Chen YL
杂志排行

World Journal of Stem Cells的其它文章
- Bone marrow mesenchymal stem cell treatment improves poststroke cerebral function recovery by regulating gut microbiota in rats
- How mesenchymal stem cell cotransplantation with hematopoietic stem cells can improve engraftment in animal models
- Combination of mesenchymal stem cells and three-dimensional collagen scaffold preserves ventricular remodeling in rat myocardial infarction model
- Sinomenine promotes differentiation of induced pluripotent stem cells into immature dendritic cells with high induction of immune tolerance
- Pancreatic transplant surgery and stem cell therapy:Finding the balance between therapeutic advances and ethical principles