ASPM基因缺陷致新生儿小头畸形6例病例系列报告并文献复习
2022-10-07俞可欣梅红芳陈辉耀张坚涛胡黎园程国强卢宇蓝王慧君吴冰冰周文浩
俞可欣 梅红芳 陈辉耀 张坚涛 胡黎园 程国强 卢宇蓝 王慧君 吴冰冰周文浩,4 杨 琳
原发性小头畸形(MCPH)又称常染色体隐性小头畸形,是一种以头围小、面部畸形和智力障碍为特征的罕见遗传性疾病。主要临床诊断标准是出生时枕额围小于同性别和胎龄新生儿2个或以上标准差,6月龄时相差达3个标准差[1]。全世界MCPH的发病率为1/250 000~1/30 000[2]。MCPH患儿通常预期寿命缩短,同时对患儿的照顾和管理也造成一定的家庭和社会负担。目前已知的MCPH相关基因中,MCPH5即异常纺锤体同源小头畸形相关(ASPM)基因报道最多。仅在巴基斯坦人群中,ASPM小头畸形(ASPM-MCPH)就占MCPH的68.8%[3]。截至目前,人类基因突变数据库(HGMD)共收录了ASPM基因255种变异,主要致病变异谱包括无义变异、移码变异、剪切位点变异。
本文回顾性分析复旦大学附属儿科医院(我院)分子医学中心通过基因检测确诊的ASPM-MCPH患儿的临床资料和基因变异情况,并行文献复习,由此拓展该疾病变异谱,深入对ASPM-MCPH的认知与探究。
1 方法
1.1 小头畸形临床诊断标准 ①可提供生后7 d内头围测量值,②枕额围小于出生时同性别、胎龄平均值2个标准差及以上[4]。
1.2 病例纳入标准 同时满足以下3项:①新生儿基因组计划(CNGP)数据库中的新生儿病例[5];②ASPM基因致病/可疑致病(P/LP)的复合杂合或纯合变异;③符合新生儿MCPH临床诊断标准。
1.3 高通量测序分析 取得患儿父母知情同意后,采集患儿外周静脉血,采用mini blood全血试剂盒(QIAGEN公司,德国)进行基因组DNA提取,NanoDrop 2000(Thermofisher公司,美国)紫外分光光度仪测定其浓度。使用Cleaseq捕获试剂盒进行基因捕获,并基于Illumina Hiseq 2000/2500平台进行序列检测。测序覆盖度>98%,平均测序深度>100×。通过高通量测序数据分析流程对变异进行信息注释和致病性分析[6]。
1.4ASPM基因P/LP变异的评级标准和携带频率评估 根据ACMG指南[7]进行致病性分析,并参考前期P/LP变异评级研究[6, 8]。P变异评级标准: 满足以下2项,①检测到该基因纯合或复合杂合变异,且可以解释先证者表型;②已经报道的明确致病变异。LP变异评级标准:同时满足以下3项中任意2项,其中必须满足第①项,①检测到该基因纯合或复合杂合变异,且可以解释先证者表型;②与已知致病变异位点相同但碱基及氨基酸改变不同;③失功能变异(无义变异、移码变异、剪接位点+/-1或2、起始密码子变异、单个或多个外显子缺失)。
鉴于在CNGP数据库中采集的ASPM基因的P/LP变异的人数局限性,故同时选择HGMD数据库(http://www. hgmd.cf.ac.uk)和ClinVar数据库(https://www.ncbi.nlm.nih.gov/clinvar)中的收录ASPM基因的P/LP变异,经由2位遗传学专家独自依照ACMG指南进行致病性判定,采用R软件计算(3.5.1,http://cran.r-project.org),CNGP数据库新生儿人群ASPM基因P/LP变异的携带频率=ASPM基因P/LP变异数(单杂合+复合杂合×2+纯杂合×2)/CNGP新生儿总例数×2。
1.5 临床资料提取 CNGP包括了我院和外院新生儿病例,我院新生儿病例从病历系统中截取如下信息:性别、胎龄、出生体重、分娩方式、产前B超检查结果、家族史、现病史、体格检查、MR结果;外院新生儿病例在报送CNGP时需要填写如上信息。
1.6 统计学方法 采用SPSS 25.0软件探讨失功能变异和错义变异对发育迟缓的影响。计数资料采用卡方检验和Fisher确切概率法进行比较。P<0.05时差异有统计学意义。
2 结果
2.1 CNGP数据库中ASPM基因P/LP变异携带频率 图1显示,在HGMD和ClinVar数据库收录的ASPM基因致病变异分别为224个位点和203个位点。2个数据库共同收录155个位点;表1显示,在CNGP数据库中携带的ASPM基因复合杂合P/LP变异12个位点。12个P/LP变异均为失功能变异,其中无义变异4个、移码变异7个、剪切位点变异1个。6个变异位于18号外显子,3个变异位于3号外显子,2个变异位于23号外显子,1个变异位于15号外显子。CNGP数据库分别与HGMD和ClinVar数据库各重复1个ASPM基因P/LP变异位点,CNGP数据库与HGMD和ClinVar数据库重复4个ASPM基因P/LP变异位点,CNGP数据库新发现6个ASPM基因P/LP变异位点。最终纳入计算携带频率的致病变异点共163个(图1中红色字)。CNGP数据库中15 754例新生儿,无纯合P/LP变异,单杂合P/LP变异26例,复合杂合P/LP变异6例。ASPM致病基因等位基因携带频率是12.060 43/万。
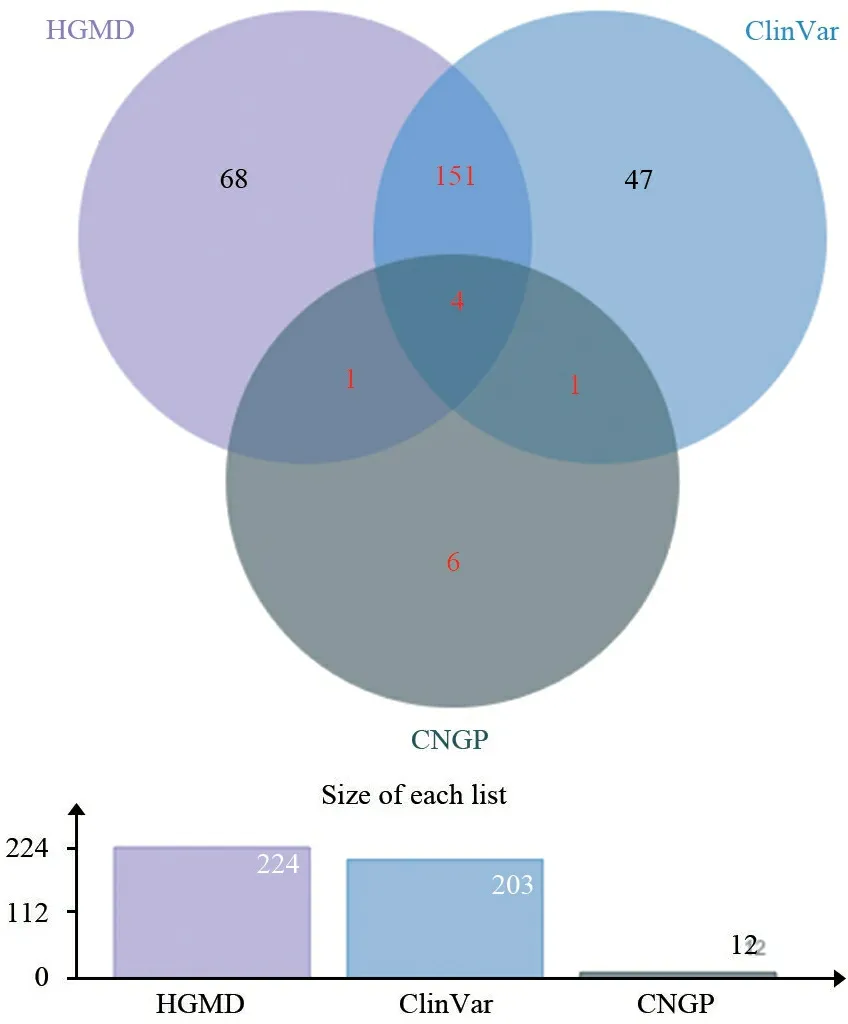
图1 ASPM基因致病变异数据库构成分布图

表1 6例新生儿ASPM基因变异位点
2.2ASPM基因复合杂合P/LP变异病例一般情况 6例复合杂合P/LP变异新生儿均表现为MCPH,符合本文纳入标准;男、女各3例,胎龄在35~39周,出生体重2.15~2.95 kg;产前B超提示:5例双顶径偏小,1例胎儿整体偏小;2例有特殊面容,1例前额突出、耳轮廓小,1例小下颌,通贯手和湿疹样皮损各1例;3例头颅MR异常表现,1例脑发育不良(脑回数量略减少、脑沟浅平、脑室扩大、脑外间隙增宽、大脑白质少);1例小头畸形伴少脑回畸形;1例颅脑发育异常伴枕部囊肿,3例头颅MR未见异常。
3 文献复习
3.1 临床表型和基因型关联性分析 选取HGMD数据库(截止到2021年4月)收录的ASPM基因的P/LP变异224个位点,来源于55篇文献报告(717例),双人独立筛选和提取临床表型和基因型,意见不一致时讨论决定。排除ASPM基因单杂合P/LP变异4篇(4例),排除未报告临床表型11篇(16例),最终对40篇文献ASPM基因的P/LP变异168个位点(697例)与临床表型行关联性分析。需要说明的是,临床表型中发育迟缓的分级判断标准参考文献[9](将智力障碍归为发育迟缓),严重发育迟缓:全智商(FSIQ)<34分或发育商(DQ)<39分,轻中度发育迟缓:FSIQ ~90分或DQ ~79分。
168个ASPM基因的P/LP变异位点中,失功能变异162个 (96.4%),包括无义变异68个,移码变异80个,剪切位点变异14个;错义变异6个。
ASPM基因的P/LP变异的697例患儿的临床表型,①头面部特征:小头畸形99.4%(624/628),前额倾斜82.4%(70/85);②发育评估:轻中度发育迟缓67.3%(233/346),严重发育迟缓20.5%(71/346),语言发育落后47.9%(90/188),运动发育落后33.7%(34/101),行为异常34.2%(77/225);③MR表现:脑回异常77.9%(81/104),胼胝体发育不良47.2%(34/72),脑室扩大50.0%(36/72),小脑和/或脑桥发育不全28.6%(20/70),脑囊肿3.4%(2/58),多小脑回10.9%(6/55);④其他罕见表型:身材矮小33.8%(45/133),色素减退和/或色素沉着12.8%(6/47),脊柱侧弯12.0%(6/50),张力减退7.6%(5/66),手脚畸形1.5%(2/133),张力亢进4.1%(2/49),斜视16.3%(8/49)。
346例发育迟缓患儿中,301例提供了严重程度的分级,其中,错义变异引起轻中度发育迟缓9例,严重发育迟缓4例;失功能变异导致轻中度发育迟缓224例,严重发育迟缓64例。在233例轻中度发育迟缓的患儿中,失功能变异的等位基因型频率为448/466,错义变异等位基因型频率为18/466;在68例严重发育迟缓的患儿中,失功能变异的基因型频率为128/136。统计分析认为失功能变异和错义变异导致的发育迟缓程度差异无统计学意义。错义变异的基因型频率为8/136。需要说明的是,由于错义变异的报道非常少,且无临床信息或未提供发育迟缓分度的病例无法列入计算,因此其结果对表型和基因型关联性的意义有限,可能存在偏倚。
4 讨论
MCPH是一种罕见遗传性疾病,由神经源性有丝分裂障碍引起。目前发现与MCPH有关的基因主要在细胞分裂中表达,其变异将导致神经发生、细胞周期检查点、中心体和纺锤体定位的中断,最终造成正常大脑体积的整体缩小,特别是在大脑皮层区域[3]。有研究新发现RRP7A[10]、TTI2[11]、LMNB1及LMNB2[12]基因的变异亦可引起小头畸形,于是MCPH相关基因在熟知的25个基因座的基础上更进一步[13],截至目前,OMIM收录的MCPH相关基因共28个。目前报道的MCPH在遗传模式上大多数呈常染色体隐性遗传,仅ALFY、LMNB1和LMNB1基因导致的小头畸形被认为呈常染色体显性遗传[14]。在小头畸形相关基因中,ASPM基因是报道最多的一个。
ASPM基因位于染色体1q31.3,包含28个外显子,其编码的蛋白质包含3 477个氨基酸[15]。ASPM是一种微管负端相关蛋白,在有丝分裂过程中以微管依赖的方式招募到纺锤体的中心周基质(PCM)。ASPM参与了所有分裂细胞的纺锤体组织、纺锤体定位和胞质分裂,该蛋白的C端末端是ASPM定位和功能所必需的[16]。在体内敲除小鼠大脑中的Aspm,导致神经发生、神经元迁移和皮质层形成的缺陷[17]。 有研究提出,因ASPM基因区域差异性表达和在增殖祖细胞亚群中受到特异性调控的特性,ASPM-MCPH患者的脑容量在皮质和底层白质皮质下灰质和小脑减少更多[18]。
ASPM-MCPH多见于近亲婚配的后代中;但近年来,不少散发的病例被报道,得益于基因检测的普及。2019年的文献分析发现,ASPM-MCPH患者大多来自巴基斯坦、沙特阿拉伯、埃及和伊朗,其中一些国家有近亲结婚的习俗,还有一些ASPM-MCPH家族来自欧洲和美洲[19]。日本[20]、越南[21]也有少量报道。我国于2011年和2020年分别报道了一例复合杂合ASPM小头畸形的病例[2,21]。既往研究发现在MCPH患者中,ASPM基因缺陷的检出率在7%~59.5%[22-24]。也有提及ASPM基因单个变异的人群携带率,如在土耳其人群中为0.001 423 6~0.003 847 2[25]。在15 754名参与CNGP的NICU新生儿人群中,ASPM基因P/LP变异携带频率是0.001 206 043,这是首次基于较大规模的人群基因组数据计算的ASPM基因的携带情况。ASPM-MCPH在我国为非常罕见的遗传疾病,本携带率计算基于NICU新生儿人群,因此致病变异的检出率可能被高估[26]。
本文通过系统整理HGMD收录的记录,有较为完整的临床信息的病例共697例。发现ASPM-MCPH患儿中,占比>50%的表型特征为:小头畸形占99.4%,特殊面容占82.35%,轻中度发育迟缓占67.3%,脑回异常占77.9%,脑室扩大占50.0%。本文报道的6例患儿,均为新生儿期诊断,尚不能评价后期发育情况。但值得注意的是,5例均在产前已提示胎儿双顶径偏小,提示对于此类宫内已明确提示头围偏小的患儿,应高度重视。2例患儿存在特殊面容,其中1例为前额突出、耳轮廓小,另1例为小下颌。3例患儿头颅MR提示异常,其中1例表现为脑回数量略减少,脑沟浅平、脑室扩大、脑外间隙增宽,大脑白质少;小头畸形伴少脑回畸形1例,颅脑发育异常伴枕部囊肿1例。2例患儿有皮损表现,其中例3腰背部、双侧臀部大片色素痣,颈部、前胸、下肢小片色素痣。既往报道中有色素减退/色素沉着斑疹表现,但较罕见[18]。病例1头部和躯干四肢较多湿疹样皮损,既往未见相关报道,是否为ASPM-MCPH罕见表现,尚无定论。既往报道中的罕见表型与MCPH的关系仍待进一步证明,目前其可能是患儿基因送检的起因,但不能作为诊断的依据之一。
失功能变异是ASPM-MCPH的主要致病变异谱。本文列入分析的168个变异,发现ASPM基因的P/LP变异中,绝大多数为失功能变异(96.43%),移码变异最多,其次为无义变异,错义变异仅6个。根据既往报道提供的病例表型,对错义变异和失功能变异引起发育迟缓严重程度进行统计学分析。统计分析认为失功能变异和错义变异导致的发育迟缓程度差异无统计学意义。不排除错义变异的病例少、既往病例信息不足导致统计分析可能存在偏倚的影响。在本文病例筛选过程中,发现双等位基因错义变异或失功能变异/错义变异的病例,患儿无典型小头畸形,但有发育迟缓或孤独症的表现,这些基因变异的评级为VUS。ASPM基因的某些错义变异对神经发育可能产生负面影响,有一定的致病性,但目前错义变异病例少,其中的机制有待进一步阐明。
ASPM基因缺陷使MCPH患儿的神经发育受到不同程度的影响,尽早明确病因有利于精确诊断、治疗开展和预后评估。本文6例患儿在产前B超中均发现头围偏小,出生后经基因检测确诊,显现出分子诊断的精准性和临床应用价值。本文病例及近年报道的散发病例有力地证明,基因诊断技术的提高和普及,有利于罕见遗传病散发病例被及时发现,及早治疗。ASPM-MCPH患儿需要个体化的对症治疗,在儿保科随访进行发育评估和行为治疗[27]。应为患儿家庭提供遗传咨询。
本研究的局限性:①ASPM基因 P/LP变异携带频率的评估在NICU人群中展开,具有人群偏倚,不能代表健康人群P/LP变异等位基因的携带水平;②既往报道中错义变异少,且既往文献对表型和基因型的描述详略不一,本文的分析结果仅有一定的参考意义。③在本文的统计分析中,未对轻、中度发育迟缓进行分类。
