紫外指纹图谱结合化学计量学对黄精基原植物的鉴别
2022-09-29杜泽飞段宝忠
杜泽飞,张 霞,温 翔,段宝忠
(1. 个旧市人民医院药剂科,云南 红河 661000;2. 大理大学药学院,云南 大理 671000)
黄精为大宗中药材,始载于《神农本草经》,为2020版《中国药典》收载品种,其法定基原为多花黄精(Polygonatum cyrtonema)、滇黄精(P.kingianum)、黄精(P. sibiricum)的干燥根茎,具有润肺滋阴、补脾益气、补肾益精之功效[1]。现代药理学研究表明,黄精主要含有多糖、皂苷类和黄酮类等成分,具有抗氧化、抗肿瘤、抗凝血和免疫调节等生物活性[2],是天然的保健食品,兼具食药用价值。黄精种类分布广泛,受地理、环境等因素影响,不同基原黄精的化学成分、营养物质含量、药效等不尽相同[3—6],对其进行鉴别分类是后续研究和开发利用的基础。但不同基原黄精形态较为相似,不易准确鉴别[7—8]。
常用的中药材鉴别方法有形态鉴别、显微鉴别、DNA 分子鉴别、色谱鉴别等[9—10],其中光谱鉴别具有稳定、简便、快速的特点,且光谱技术愈发成熟,已广泛用于植物分类和中药材种类鉴别[11—13]。不同物质体系成分的不饱和程度不同,紫外指纹图谱技术中的紫外吸收光谱曲线峰形、峰高、峰面积等均有一定的差异[14]。通过比较图谱不同吸收峰的差异,可对药材种类快速简便区分[15],如中药灵芝及其伪品的有效鉴别[16]、不同产地的中药三七鉴别[17]、不同产地和种类的牛肝菌鉴别[18—20]。目前,尚未见黄精法定基原植物的紫外光谱鉴别研究。本研究对3种法定基原黄精进行紫外光谱分析,并结合多元统计学,包括主成分(PCA)和系统聚类分析(HCA)快速鉴别黄精基原种,为黄精临床应用和质量评价提供借鉴。
1 仪器、试剂及材料
1.1 仪器试剂
UV-9000型紫外可见分光光度仪(上海元析仪器有限公司)、AL204型电子天平(梅特勒-托利多仪器上海有限公司)、高温干燥箱(上海-恒科学仪器有限公司)、中药粉碎机(佛山市德玛仕网络科技有限公司)、超声波清洗仪(宁波新芝生物科技有限公司)、100目不锈钢筛(宁波新芝生物科技有限公司)。氢氧化钠、甲醇、无水乙醇均为分析纯,水为超纯水。
1.2 材料
药材样品采购于云南、安徽等地(表1),经大理大学段宝忠教授鉴定为百合科黄精属植物黄精(S1~S9)、滇黄精(S10~S21)和多花黄精(S22~S33)的新鲜根茎,所有样品干燥后粉碎,备用。
2 方法
2.1 黄精紫外光谱测试液制备
黄精样品粉碎过100目筛备用。称取0.30 g样品置于 10 mL无水乙醇溶液中,室温下超声提取30 min,过滤得黄精紫外光谱测试液。
2.2 黄精紫外光谱测定
测定波长190~400 nm,狭缝1.0 nm,采样间隔0.2 nm,重复3次。以无水乙醇作为参比液和测试液进行基线校正及空白测定。
2.3 黄精样品提取条件单因素实验[18—20]
2.3.1 提取溶剂
随机选取S1样品,准确称取0.10 g样品于10 mL具塞试管中,分别加入蒸馏水、无水乙醇、甲醇和0.5 mol·L-1氢氧化钠各10 mL,每组平行3次;超声提取30 min,过滤,以对应的溶剂为参比液,测定紫外光谱,根据吸收峰数确定最佳提取溶剂。
2.3.2 提取时间
称取S1样品0.10 g,加10 mL无水乙醇,分别超声提取20、30、40、50、60 min,以溶剂为参比液,扫描紫外光谱,根据吸收峰数确定适宜提取时间。
2.3.3 提取用量
称取S1样品0.10、0.15、0.2、0.25、0.3 g,分别加入10 mL无水乙醇,超声提取30 min。以溶剂为参比液,测定紫外光谱,根据吸收峰数确定最佳样品用量。
2.4 方法学考查
2.4.1 精密度
称取S1样品0.3 g样品1份,按照2.2的方法重复测定6次,计算最大吸收波长(210 nm)处的变异系数RSD(%),考查精密度。
2.4.2 重复性
称取S1样品6份,每份0.3 g,按照2.2的方法测定,计算最大吸收波长(210 nm)处的变异系数RSD(%),考查重复性。
2.4.3 稳定性
称取S1样品0.3 g,按照2.1的方法提取,分别在放置1、5、10、20、30 h时测定紫外光谱,计算最大吸收波长(210 nm)处的变异系数RSD(%),考查稳定性。
2.5 数据处理
取波长190~450 nm范围内的紫外光谱数据,转置后用SPSS 20.0软件进行聚类分析与主成分分析,直观表征不同基原黄精样品间的相似性。
3 结果与分析
3.1 黄精特征成分提取条件的优化
3.1.1 最优提取溶剂
图1A为相同条件下不同溶剂提取样品S1的紫外指纹图谱,根据图谱吸收峰数确定不同溶剂对黄精提取率的影响。结果显示,相同条件下蒸馏水、甲醇和 0.5 mol·L-1氢氧化钠提取液有 1~3个吸收峰;无水乙醇提取液在190~300 nm波长范围有明显的紫外吸收峰,共有5个吸收峰,吸收峰数目较多。因此,用无水乙醇作为提取溶剂。
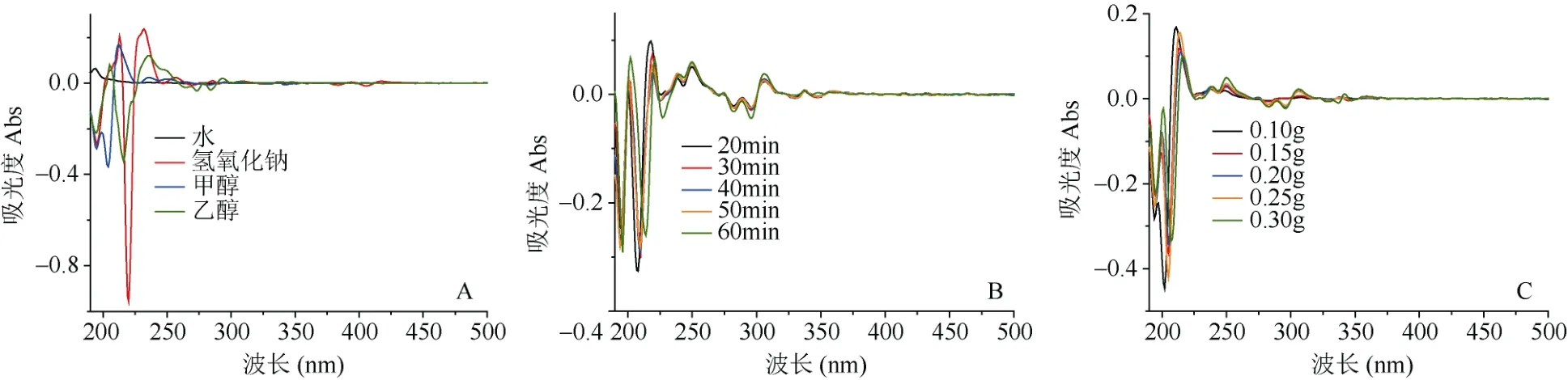
图1 不同提取条件下黄精样品紫外指纹图谱Fig. 1 UV fingerprint of polygonatum samples under different extraction conditions
3.1.2 最佳提取时间
以无水乙醇作为提取溶剂,考查不同提取时间对提取效果的影响。从图1B可看出,提取20 min时,黄精样品提取液的紫外吸收峰较少,只有5个,集中在波长190~350 nm范围内;当提取30 min时,吸收峰数目较多,共8个;当提取60 min时吸收峰数目没有明显变化。故提取时间30 min较宜。
3.1.3 最适称样量
以无水乙醇为提取溶剂,研究不同称样量对黄精紫外光谱的影响。结果表明,黄精样品取0.1 g时,提取液的紫外吸收峰较少,只有3个吸收峰;当用量为0.3 g时,吸收峰较多,共7个(图1C)。故样品用量取0.3 g较好。
3.2 试验方法考查
黄精样品无水乙醇提取液的紫外光谱的重复性变异系数RSD在0.03%~0.63%之间,精密度的变异系数RSD介于0.00~2.87%之间,30 h内稳定性的变异系数RSD在0.04%~1.73%之间,表明该方法精密度高、重现性好,稳定可靠。
3.3 黄精紫外指纹图谱对比分析
图2为不同种类黄精的紫外指纹图谱,图2A为3种黄精平均紫外光谱图,图2B为3种黄精一阶倒数处理紫外光谱图。由图2A可以看出,不同种类黄精的紫外指纹图谱在波长200~400 nm范围内主要紫外吸收峰出现的位置基本一致,如210 nm、280 nm等处是黄精样品的共有峰,表明不同基原黄精主要化学组分相似;但不同样品的吸光度差异较为明显,如多花黄精在210 nm和281 nm 等处的吸光度较高,吸光度值分别为2.74和1.24,而黄精在这两个波长下的吸光度为2.72和0.92,滇黄精则为2.50和0.80(图2A),多花黄精、黄精、滇黄精的吸光度A210: A281分别为2.21、2.95、3.13,滇黄精的吸光度比值最大,而多花黄精的吸光度比值最小,不同黄精样品表现出指纹性和特征性,表明3种黄精化学成分的含量存在一定的差异,这可能是因种类、产地、生长环境不同导致的成分含量或化学成分构型种间差异。由图2B可以看出,黄精和多花黄精在210 nm处有吸收峰,且黄精的吸收峰较强,但滇黄精在此附近无吸收峰,在220 nm与280 nm附近多花黄精的吸收较强。因此,利用紫外波长210 nm、220 nm、280 nm吸收区域或不同波长下吸光度值的比值可进行黄精基原植物鉴别。
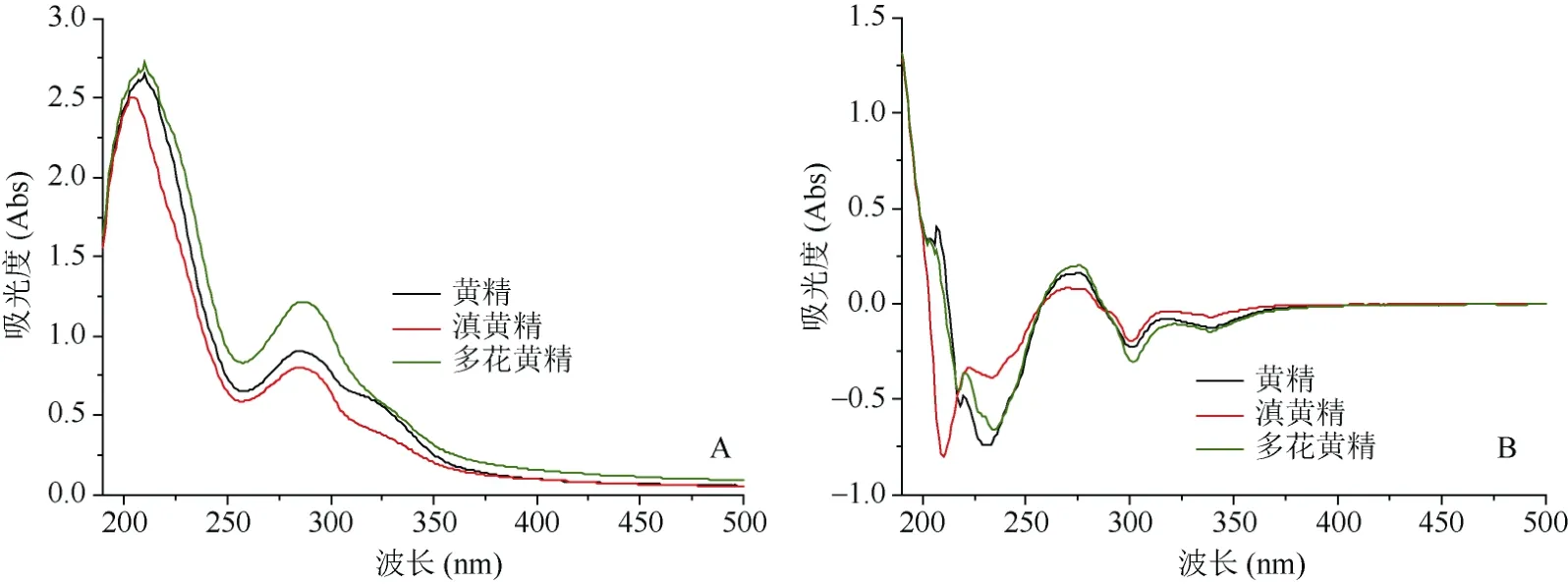
图2 黄精样品紫外指纹图谱Fig. 2 UV fingerprint of three Polygonatum species
3.4 HCA分析
采用平方欧式距离,应用系统聚类分析(HCA)中的Ward聚类法对3种基原黄精进行分析(图3)。33批黄精药材在分类距离为25时,被分为两大类,Ⅰ类包括12批滇黄精样品(S10~S21),Ⅱ类包括9批黄精和11批多花黄精。滇黄精聚为一支,与黄精和多花黄精明显区分。根据聚类分析,滇黄精和其他两种黄精的化学成分存在显著差异。而黄精和多花黄精具有相似的化学成分,无法通过聚类分析进行区分。

图3 33批黄精样品HCA树状图Fig. 3 The cluster analysis of Polygonatum samples
3.5 PCA分析
将3种黄精紫外光谱全波段谱图进行一阶导数处理后,用SPSS20.0软件进行主成分分析(PCA)。PCA提取的前3个主成分作为坐标轴,绘制3种黄精的主成分分析三维得分图。主成分得分图反映不同种类黄精紫外指纹图谱间的关系,图谱相似的样品在主成分得分图中聚在较近的区域,而图谱差异越大在主成分得分图中距离越远。研究表明,前三个主成分累计贡献率达98.36%,能够表达黄精紫外光谱的大量信息,其中第一主成分特征值为27.79,方差贡献率为 84.21%,第二主成分特征值为 3.8,方差贡献率为11.51%,第三主成分特征值为0.87,方差贡献率为2.64%。由图4可见,33批黄精药材被分为2大类,其中滇黄精样品聚为一类(Ⅰ),黄精和多花黄精样品聚为另一类(Ⅱ),与聚类分析结果一致。主成分分析能够反映不同种类黄精的组分和含量差异,根据主成分分析结果可以鉴别不同种类黄精。
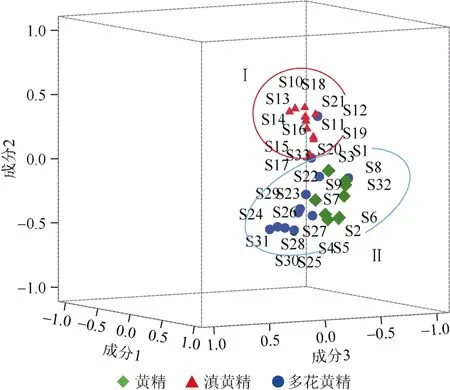
图4 33批黄精样品PCA图Fig. 4 PCA results of Polygonatum samples
4 讨论与结论
黄精为多元道地药材,市场上除了3种法定基原黄精外,其同属植物也均有流通,但传统认为黄精质量最佳。2020年版《中国药典》鉴定项目中仅有黄精性状的描述及多糖含量的测定,但这两项难以准确鉴定其种类。本研究发现,3种黄精基原植物的紫外指纹图谱聚类分析中,滇黄精单独聚为一支,与黄精和多花黄精明显区分,与采用黄精多糖的差异性区分基原黄精的结果一致[6]。笔者前期采用红外光谱结合化学计量学可将3种基原黄精加以区分[21]。杨兴鑫等[4]采用UPLC-Orbitrap MS技术分别对黄精化学成分进行表征,发现3种法定基原黄精明显分为3类。目前,采用紫外光谱方法对黄精基原植物进行鉴别未见报道,或因紫外光谱数据易受测试条件如温度、湿度、溶剂等的影响,所测数值为多成分共同吸收,又以主成分的影响为主,对细微成分区别度较低。
黄精化学成分构成复杂,不同提取条件对黄精图谱显示结果有较大差异。本实验通过对黄精提取条件优化,建立33个不同基原黄精的平均紫外指纹图谱,不同种类黄精图谱差异明显。三种黄精紫外光谱在波长210 nm、220 nm、280 nm附近差异明显,其共有峰揭示了组成成分的相似,而吸光度差异反映出含量不同。聚类分析和主成分分析显示,不同种类黄精对物质的积累具有差异,这可能与黄精的生长环境、采收季节等因素有关;紫外光谱结合主成分分析可以区分滇黄精与黄精、多花黄精样品,可为不同基原黄精鉴别和质量控制提供参考。
