响应面法优化硒化桑叶多糖的制备工艺及其体外抗氧化活性
2022-09-24胡润锋李浚哲李鹏飞李湘洲
胡润锋,李浚哲,李鹏飞,周 军,李湘洲
(1.中南林业科技大学 材料科学与工程学院,湖南 长沙 410004;2.广西民族大学a.广西林产化学与工程重点实验室;b.广西林产化学与工程协同创新中心,广西 南宁 530006)
桑Morus alba是桑科、桑属落叶乔木或灌木,广泛分布在温带、亚热带、热带等气候地区。我国是世界上最大的种桑国,已有4000 多年的桑栽培史。桑叶为植物桑的干燥叶,又名铁扇子,神仙叶。现代药理学研究表明,桑叶因含黄酮、生物碱、甾醇、γ-氨基丁酸、多糖等多种天然活性成分[1-2],使其具有降血糖[3]、抗肿瘤[4]、抗炎[5]、抗氧化[6]等多种生物活性,已被广泛应用于食品、医药及保健行业[7-8]。2002年,我国就将桑叶列为药食同源。
硒是维持人体正常生理活动所必需的18 种微量元素之一,它在维持机体正常功能和健康方面有着不可或缺的地位[9]。研究表明,硒元素可增强体内抗氧化能力、降低癌症风险、提高机体免疫力,而硒元素的缺乏也与肝损伤,心血管疾病,癌症和衰老等有关[10-11]。因此,对于缺硒人群来说,硒元素的补充尤为重要。硒化合物按照硒元素的形态可分为有机硒化合物(硒多糖、硒氨基酸、硒蛋白)和无机硒化合物(硒酸盐、亚硒酸盐),相比于无机硒化合物,有机硒化合物具有生物利用度高和生物毒性低等特点,更适合用于补充人体的硒元素,其中富硒多糖便是一种理想的补硒制剂[12]。硒多糖可分为天然硒多糖和人工合成硒多糖。天然硒多糖主要存在于富硒土壤中生长的植物内以及用富硒培养基培养的微生物内[13-15],但由于富硒土壤分布不均匀以及植物和微生物的富硒能力有限,导致天然富硒产品产量少且硒多糖含量较低[16]。人工合成硒多糖是由天然多糖和无机硒化合物合成得到的,相比于天然硒多糖,具有原料来源丰富且硒含量高的特点,兼具多糖和硒元素两者的生物活性[17-18]。近年来,国内外学者利用天然多糖和无机硒化物反应成功制备出各种富硒多糖,不仅保留甚至提高了天然多糖原有的生物活性,同时也增加了硒元素独特的的生物活性,且毒副作用大大降低。Yuan 等[19]利用微波辅助技术合成的硒化甘薯多糖可以显著提高甘薯多糖的体外抗氧化、体内抗肿瘤、降血糖活性。Huang 等[20]合成的硒化黄芪多糖提高了黄芪多糖的清除自由基和抑制肾结石的活性。Liu 等[21]合成的硒化梭柄乳头菌多糖的降血糖、降血脂活性明显提高。目前,将硒元素引入桑叶多糖以提高桑叶多糖的生物活性还未见过报道。
桑叶多糖作为桑叶的主要活性成分之一,具有降血糖、抗氧化、抗凝血等生物活性[22-23],而硒元素同样具有显著的抗氧化活性,将两者结合能否提高桑叶多糖的抗氧化活性有待探究。值得注意的是,在硒化多糖改性过程中,反应条件能对多糖的硒含量造成较大的影响,而硒含量能直接影响到硒化多糖的生物活性,因此对天然多糖的硒化反应条件进行优化具有重要意义。基于此,本研究以桑叶多糖为原料,硒含量为考察指标,利用HNO3-Na2SeO3法制备硒化桑叶多糖,在单因素实验的基础上利用响应面法优化硒化桑叶多糖的制备工艺,并对比研究硒化前后桑叶多糖的结构、理化性质和体外抗氧化活性的差异,为桑叶中多糖类化合物在功能性食品和药物等领域的开发与利用提供依据。
1 材料与方法
1.1 材料与仪器
干桑叶,产自湖南永州;亚硒酸钠,西陇科学股份有限公司;氯化钡、无水硫酸钠、乙二胺四乙酸二钠、无水乙醇、丙酮、乙醚、高氯酸、硝酸、盐酸、邻苯二胺、氨水、甲苯,溴化钾均为分析纯,国药集团化学试剂有限公司;S-8 大孔树脂、聚酰胺树脂,浙江省台州市路桥四甲生化塑料公司。
DF-101S 集热式恒温加热磁力搅拌器,巩义市予华仪器有限公司;WB-2000 旋转蒸发仪,郑州长城科工贸有限公司;TG16-WS 离心机,长沙平凡仪器仪表有限公司;DK-98-Ⅱ电炉,天津市天泰有限公司;Zeiss Sigma 300 扫描电子显微镜,卡尔·蔡司股份公司;Agilent 1260 Infinity 高效液相色谱仪,安捷伦科技有限公司;Zetasizernano 纳米粒度及zeta 电位分析仪,英国马尔文仪器有限公司;ALPHA 傅里叶变换红外光谱仪,布鲁克公司;UV2300-Ⅱ系列双光束紫外可见分光光度计,上海天美科学仪器有限公司。
1.2 实验方法
1.2.1 MLP-Se 的合成
利用水提醇沉法提取桑叶多糖[24]。称取一定量粉碎烘干后的桑叶粉,按照固液比1∶20(g·mL-1)加入无水乙醇,80℃回流2 h 后过滤,滤渣60℃烘干得到脱脂桑叶粉。称取一定量脱脂桑叶粉,按照固液比1∶20(g·mL-1)加入去离子水,80℃回流提取2 h,离心,取上清液。依次用聚酰胺树脂和S-8 大孔树脂动态吸附法除去提取液中蛋白和色素,浓缩后加入4 倍体积的无水乙醇,5℃下静置24 h,过滤,沉淀依次分别用无水乙醇、丙酮、乙醚洗涤3 次,冷冻干燥得到桑叶多糖,命名为MLP。
利用HNO3-Na2SeO3法对MLP进行硒化改性。称取300 mg 的MLP 置于锥形瓶内,加入50 mL一定质量浓度的HNO3,加热使其充分溶解,加入一定量的Na2SeO3与700 mg 的BaCl2,水浴加热搅拌反应一定时间。反应结束后使其冷却,调节pH 值至7 左右,加入一定量的Na2SO4以除去BaCl2,离心取上清液置于透析袋中透析72 h,浓缩、冻干后得到硒化桑叶多糖,命名为MLP-Se。桑叶多糖硒化的反应方程式见图1。
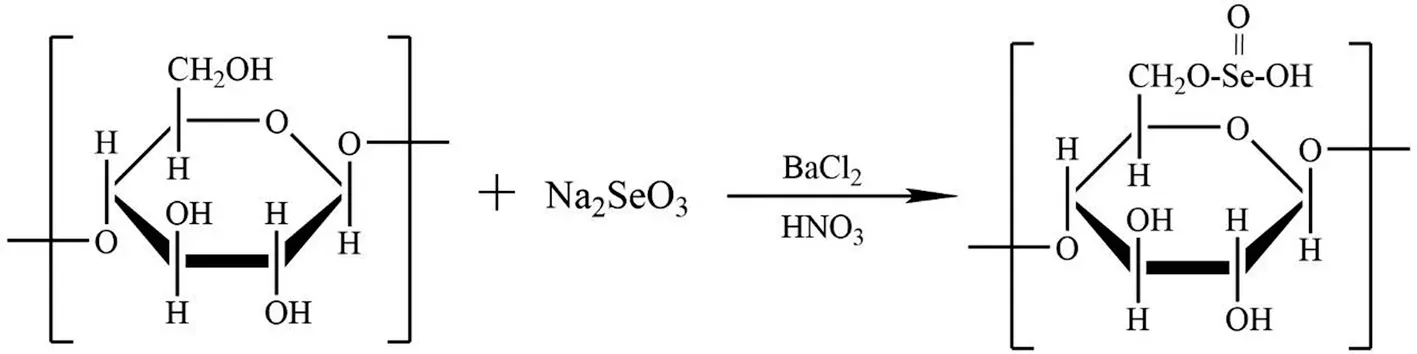
图1 桑叶多糖硒化的反应方程式Fig.1 Reaction equation of selenization of mulberry leaves polysaccharides
1.2.2 硒含量标准曲线的建立及测定
采用邻苯二胺法测定MLP-Se 中的硒含量[25]。用Na2SeO3配置一系列质量浓度梯度的硒标准溶液,定容至25 mL,加入2 mL 质量浓度2%的邻苯二胺溶液,暗处反应1 h,加入10 mL 甲苯,振荡萃取5 min,收集上层甲苯层溶液,以甲苯作为参比溶液,在波长334 nm 的紫外分光光度计中测定并得到吸光度值。以硒的质量浓度为横坐标、吸光度值为纵坐标绘制标准曲线,得到标准曲线为Y=0.307 3X+0.009 3(R2=0.998 0)。
MLP-Se 消化过程如下:称取10 mg MLP-Se置于烧杯内,加入5 mL 混合酸(HClO4∶H2SO4∶HNO3=1∶1∶4,v/v/v),浸泡3 h,待MLP-Se 完全溶解,加热消化使溶液体积蒸发至约1 mL。待消化液冷却后加入5 mL 的6 mol·L-1盐酸继续加热30 min 以上并不再沸腾时,停止加热使Se(VI)还原为Se(IV),加热过程中防止暴沸。溶液冷却后转移至25 mL 容量瓶中,加入2 mL 2%的邻苯二胺溶液和5%的EDTA-2Na 溶液,用氨水调节pH值至2.0,置于暗处反应1 h,后续操作按照标准曲线的测定方法,得到吸光度值并代入标准曲线计算硒含量。
1.2.3 单因素实验
按照MLP-Se 的合成方法,分别考察反应时间、反应温度、Na2SeO3与MLP 的质量比和HNO3的体积分数对制备的MLP-Se 中Se 含量的影响。其中,在Na2SeO3与MLP 的质量比1.2、HNO3体积分数0.5%和反应温度70℃的条件下,考察反应时间分别为5、6、7、8、9 h 对MLP-Se 中Se 含量的影响;在HNO3体积分数0.5%、反应温度80℃和反应时间7 h 的条件下,考察Na2SeO3与MLP的质量比分别为0.6、0.8、1.0、1.2、1.4 对MLPSe 中Se 含量的影响;在Na2SeO3与MLP 的质量比1.2、反应温度80℃和反应时间7 h 的条件下,考察HNO3体积分数分别为0.1%、0.3%、0.5%、0.7%、0.9% 对MLP-Se 中Se 含量的影响;在Na2SeO3与MLP 的质量比1.2、HNO3体积分数0.5%和反应时间7 h 的条件下,考察反应温度分别为50、60、70、80、90 ℃对MLP-Se 中Se 含量的影响。
1.2.4 响应面优化实验设计
在单因素实验的基础上,固定反应时间7 h,进一步优化Na2SeO3与MLP 的质量比、HNO3的体积分数、反应温度对MLP-Se 合成的影响,采用三因素三水平的Box-Behnken 方法进行优化设计(表1)。

表1 Box-Behnken 设计因素水平及编码值Table 1 Design factor level and code value of Box-Benhnken
1.2.5 扫描电镜-能谱分析
分别取干燥的MLP 和MLP-Se,粘附到碳导电介质板上镀金。喷金后将样品置于扫描电镜下室温扫描,观察硒化前后多糖的表面形貌,并利用EDS 分析仪进行元素分析。
1.2.6 凝胶色谱分析
采用凝胶色谱法测定MLP 和MLP-Se 的分子量[26]。色谱条件:Agilent 1260 Infinity 高效液相色谱仪,PL aquagel-OH(8.0×300 mm,8 μm)柱,示差折光器(RID),柱温35℃,流动相为0.1 M的NaCl 溶液,流速0.6 mL·min-1,进样20 μL。
以分子量3 000~200×105Da 的葡聚糖为标准品,以保留时间为横坐标,分子量的对数值(lgMw)为纵坐标作图,得到多糖分子量的标准曲线Y=-0.430 4X+9.910 7(R2=0.999 6)。将样品的色谱分析结果中的保留时间带入标准曲线计算样品的分子量。
1.2.7 粒径和zeta 电位分析
取适量MLP 和MLP-Se 溶于水中,配置成浓度为0.1 mg·mL-1的多糖溶液,在25℃的条件下,测量多糖样品的电势和粒径分布。
1.2.8 红外光谱分析
利用KBr 压片法对MLP 和MLP-Se 进行压片制样,样品在红外光谱下扫描,扫描频率64 Hz,扫描32 次,扫描范围400~4 000 cm-1。
1.2.9 清除1,1-二苯基-2-三硝基苯肼(DPPH)自由基能力的测定
取2 mL 不同浓度的样品溶液分别加入2 mL 0.2 mmol·L-1DPPH-乙醇溶液,混合均匀后室温下避光反应30 min,离心,取上清液于517 nm 处检测其吸光度值,以相应的乙醇溶液为对照。DPPH自由基清除率的计算公式为:

式(1)中:D为DPPH 自由基清除率,Ai是样品与DPPH 反应后的吸光度,Aj是无水乙醇代替DPPH 的吸光度,A0是蒸馏水代替样品的吸光度。
1.2.10 清除2,2-联氮-二(3-乙基-苯并噻唑-6-磺酸)二铵盐(ABTS)自由基能力的测定
将7.0 mmol·L-1的ABTS 溶液 与5.0 mmol·L-1的过硫酸钾溶液等体积混合,避光室温放置12 h后,加蒸馏水稀释50 倍,使其在波长734 nm 处吸光度为0.70±0.02。取0.5 mL 不同浓度的样品溶液,加4.0 mL 稀释后的ABTS 溶液,室温避光反应6 min,在734 nm 处测其吸光度,以相应蒸馏水为对照。ABTS 自由基清除率的计算公式为:

式(2)中:Abt为ABTS 自由基清除率,Ai是样品与ABTS 反应后的吸光度,Aj是蒸馏水代替ABTS的吸光度,A0是蒸馏水代替样品的吸光度。
1.2.11 数据处理与分析
实验数据用3 次独立重复实验结果的平均值±标准差表示,采用SPSS 18.0 软件进行单因素方差分析,P<0.05 为差异显著,采用Origin 9.1软件绘图。
2 结果与分析
2.1 单因素实验
按照1.2.3 中所述方法分别考察了反应时间、反应温度、Na2SeO3与MLP 的质量比和HNO3体积分数对MLP-Se 中硒含量的影响,结果见图2。
由图2a 可知,随着反应时间的延长,MLP-Se的硒含量先增大后减小,反应时间为7 h 时的硒含量达到最大值为2.08±0.06 mg·g-1。因此选择反应时间为7 h。由图2b 可知,随着反应温度的增大,MLP-Se 的硒含量先增大后减小,当反应温度为80℃时,硒含量达到最大值为2.83±0.07 mg·g-1,继续增大反应温度,硒含量开始减小,原因在于高温能激活反应位点,有利于硒化反应的进行,但过高的温度可能会导致部分多糖链断裂以及有机硒稳定性结构被破坏造成的[27]。由图2c 可知,随着HNO3体积分数的增大,MLP-Se 的硒含量先增大后减小,当HNO3体积分数为0.5%时,硒含量达到最大值为2.85±0.05 mg·g-1,继续增大HNO3体积分数,硒含量开始减小,原因可能是因为在合适的酸性条件下,糖苷键的部分断裂暴露出更多的硒化反应位点,但反应体系酸性环境过大将导致多糖结构被破坏[28]。由图2d 可知,随着Na2SeO3与MLP 质量比的增大,MLP-Se 的硒含量先增大后减小,在质量比值为1.2 时,硒含量达到最大值为2.84±0.08 mg·g-1,质量比继续增大,硒含量开始减小,原因可能是饱和结合位点导致的非特异性吸收有关[27]。
2.2 响应面试验结果与分析
按照表1的响应面试验设计进行实验,得到实验结果见表2。
以硒含量为响应值,利用Design-Expert 10.0.7软件,将表2中试验数据进行响应面分析,得到二次多项式回归方程:Y=-41.91+4.51A+0.38B+3 0.25C+0.18AB+4.92AC+0.03BC-8.75A2-0.003B2-6.04C2,并对此设计的模型进行方差分析,结果见表3。
由表3可知,模型中A、B、C、A2、B2、C2项均为极显著(P<0.01)。根据F值可知,各因素对桑叶多糖的硒含量影响大小顺序为:C(HNO3体积分数)>B(反应温度)>A(Na2SeO3与MLP 质量比)。试验所建立的模型(P<0.01)为极显著,失拟项(P>0.05)为不显著,说明所建立的响应面模型用来优化MLP-Se 制备的最佳工艺条件是可行的。信噪比36.695>4,说明该模型可预测试验结果,而修正判定系数RAdj=0.989 0,表示可以用此模型来预测98.9%的响应值。判定系数R2Pred=0.956 8,说明此模型拟合程度良好,可以利用模型来分析和预测制备的MLP-Se 的硒含量值,R2Pred=0.956 8 与R2=0.995 2 的值接近,说明该响应面设计合理。
经过Design-Expert 软件计算获得制备MLPSe 的响应面优化工艺为:Na2SeO3与MLP 的质量比1.268,HNO3的体积分数0.54%,反应温度84.98 ℃,此条件下的硒含量的预测值为3.158 mg·g-1。验证实验中,在质量比MLP 与Na2SeO3的1.27、反应温度85℃、HNO3体积分数0.54%的条件下,5 次平行实验制备的MLP-Se 中硒含量的实际平均值为3.147 mg·g-1。实验值与预测值接近,表明响应面优化设计合理、可靠。
2.3 扫描电镜-能谱分析
图3a—c 分别为MLP 和MLP-Se 的扫描电镜及MLP-Se 的能谱分析结果。由图3a 可知,MLP表面整体比较光滑,且存在不规则的、致密的沟壑,这可能是由于样品经冷冻干燥后水分的升华而导致开裂,而图3b 中MLP-Se 的表面凹凸不平,主要以无定形的结构聚集存在,表明硒化修饰改变了桑叶多糖的表观形貌。MLP-Se 的能谱分析表明C、O、Se 原子比分别为45.34%、54.07%、0.59%,Se 元素的存在表明硒化修饰的成功。

图3 MLP(a)、MLP-Se(b)的扫描电镜和MLP-Se 的能谱分析(c)Fig.3 Analysis of SEM of MLP(a),MLP-Se(b) and EDS of MLP-Se (c)
2.4 凝胶色谱分析
MLP 和MLP-Se 样品的凝胶色谱分析结果见图4。MLP 和MLP-Se 的保留时间分别为8.338和8.209 min,相对分子质量分别为2.099×103和2.385×103kDa,表明MLP 经硒化修饰后,其分子量有所增大,这是由于硒化反应过程中含硒官能团取代了原多糖分子中的羟基所致。
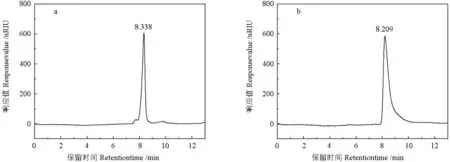
图4 MLP(a)和MLP-Se(b)的GPC 分析Fig.4 GPC analysis of MLP(a) and MLP-Se(b)
2.5 粒径和zeta 电位分析
多糖的流体力学性质如电势电位和粒径分布可以在一定程度上反映其稳定性,一般来说,多糖分子的粒径越小、zeta 电位的绝对值越大,说明多糖分子在体系中越容易分散或溶解,反之则多糖分子越容易聚集。图5为0.1 mg·mL-1的MLP和MLP-Se 溶液的粒径分布和zeta 电位分析结果,图5a 中MLP-Se 分子的平均直径(33.68 nm)小于MLP 分子的平均直径(168.0 nm),图5b 中MLP 和MLP-Se 均带负电荷,而MLP-Se 的zeta电位的绝对值(20.4 mV)大于MLP 的zeta 电位的绝对值(14.5 mV),说明硒化修饰后提高了桑叶多糖在溶液体系中的稳定性。

图5 MLP 和MLP-Se 的粒径分析(a)和zeta 电位(b)Fig.5 The particle size(a) and zeta potential distribution(b) of MLP and MLP-Se
2.6 红外光谱分析
MLP 和MLP-Se 的红外光谱分析结果见图6。由图6可知,MLP 和MLP-Se 的谱图整体相似,说明硒化修饰并不会破坏桑叶多糖的主要结构。在3 400和2 920 cm-1处的特征吸收峰分别是由O-H的弯曲振动和C-H 的伸缩振动引起的。1 625 和1 425 cm-1处为羧基的特征吸收峰,说明了糖醛酸的存在。在1 000~1 200 cm-1处的重叠峰是吡喃环中C-O-C 和C-O-H 的伸缩振动峰,说明MLP主要以吡喃糖的形式存在。而MLP-Se 分别在609、822、920 和1 384 cm-1处产生了新的特征吸收峰,其中,609 cm-1归因于Se-O-C 的不对称伸缩振动,822 cm-1归因于Se=O 伸缩振动,920 cm-1归因于C-O-Se 的伸缩振动,1 384 cm-1为糖环上与Se=O 相邻的C-H 特征吸收峰,说明MLP 经HNO3-Na2SeO3法可以有效制备MLP-Se,且硒与多糖是通过C-O-Se 和Se=O 结合的。这与Wu 等[29]和Zhu 等[30]在硒化修饰多糖的研究结果一致。

图6 MLP 和MLP-Se 红外光谱分析Fig.6 FT-IR spectroscopy analysis of MLP and MLP-Se
2.7 DPPH 自由基清除能力测定
由图7可知,MLP 和MLP-Se 对DPPH 自由基的清除率均表现出剂量依赖关系,随着样品浓度的增加,DPPH 自由基清除率越高。在相同浓度下,MLP-Se 对DPPH 自由基清除率均明显高于MLP(P<0.05)。在浓度达到5 mg·mL-1时,MLP和MLP-Se 对DPPH 的清除率分别达到70.61%和86.71%,IC50值分别为2.15 和0.88 mg·mL-1,说明硒化修饰可以增加桑叶多糖对DPPH 自由基的清除效果。

图7 MLP 和MLP-Se 对DPPH 自由基的清除能力Fig.7 Scavenging effects of MLP and MLP-Se on DPPH free radical
2.8 ABTS 自由基清除能力测定
由图8可知,MLP 和MLP-Se 对ABTS 自由基的清除率均表现出剂量依赖关系,随着样品浓度的增大,对ABTS 自由基的清除率越高。在相同浓度下,MLP-Se 对ABTS 自由基清除率的作用均高于MLP。在浓度达到5 mg·mL-1时,MLP和MLP-Se 对DPPH 的清除率分别达到80.50%和100%,IC50值分别为1.79 和0.68 mg·mL-1,说明硒化修饰可以增加桑叶多糖对ABTS 自由基清除率的作用。
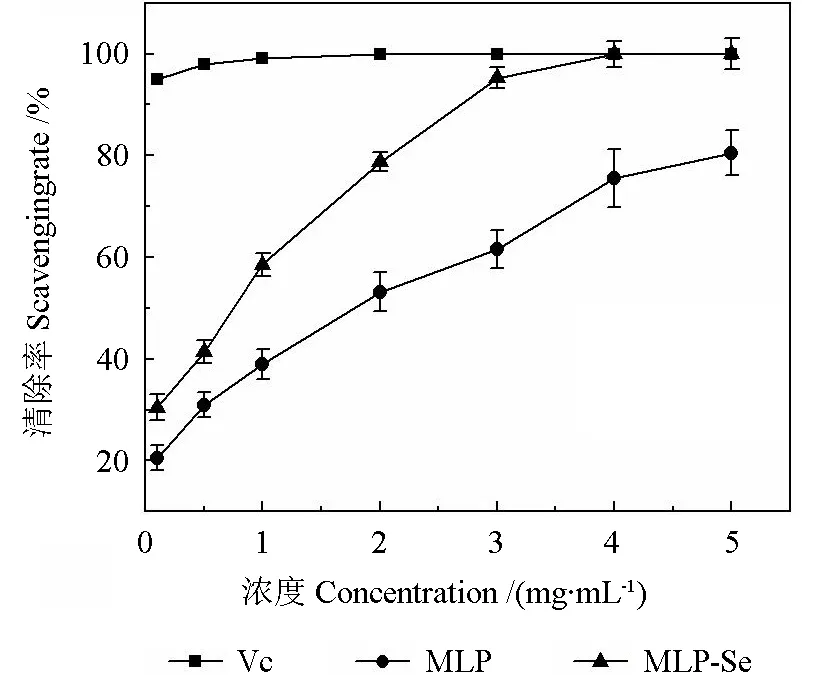
图8 MLP 和MLP-Se 对ABTS 自由基的清除效果Fig.8 Scavenging effects of MLP and MLP-Se on ABTS free radical
综上所述,利用HNO3-Na2SeO3法对桑叶多糖进行硒化改性,可以提高桑叶多糖的抗氧化活性。研究表明多糖的中羟基解离能较弱[31],其易向自由基提供氢原子,因此多糖具有抗氧化活性,且活性的强弱直接和供氢能力相关,而硒多糖中硒基团的存在可以激活正构碳中的氢原子,从而增强其清除自由基的能力[32]。
3 结论与讨论
3.1 结 论
以水提醇沉、吸附法制备的桑叶多糖(MLP)为原料,利用HNO3-Na2SeO3法对其进行硒化修饰制备硒化桑叶多糖(MLP-Se)。通过单因素和响应面法优化并获得了MLP-Se 的制备工艺为MLP与Na2SeO3的质量比1.27,反应温度85℃,HNO3体积分数0.54%,反应时间7 h,此条件下制备的MLP-Se 中硒含量高达3.147 mg·g-1,建立的响应面模型可靠。理化性质与表观结构分析表明,桑叶多糖已被成功硒化,且硒与多糖是通过C-OSe 和Se=O 结合,此外,硒化修饰改变了桑叶多糖的表面形貌,增大了桑叶多糖分子量,提高桑叶多糖在溶液体系中的稳定性。MLP-Se 对DPPH自由基和ABTS 自由基的IC50值分别为0.88 和0.68 mg·g-1,且明显低于MLP,表明硒化改性制备的MLP-Se 的抗氧化能力更强。
3.2 讨 论
将硒元素引入到桑叶多糖的分子结构中,形成富硒的桑叶多糖,不仅提高了桑叶多糖复溶后的稳定性,还有效提高了桑叶多糖原有的生物活性,为桑叶多糖的深加工开发与利用提供理论基础,也为类似天然多糖的研究与利用提供有益借鉴。通过能谱和红外光谱分析可以初步表明硒元素已成功引入到多糖分子中,且硒与多糖是通过C-O-Se 和Se=O 的方式结合的,但由于尚未对MLP 和MLP-Se 进行进一步的分离富集,因而无法对多糖修饰前后的结构进行深入解析。下一步可以在分离获得纯度较高的化合物的基础上通过圆二色谱、核磁共振波谱等分析技术对MLP 和MLP-Se 的化学结构进行深入系统的分析,以便于更好地阐述其构效关系。
