海洋沉积物孔隙水中痕量金属元素的测试分析方法*
2022-09-21王小静刘洪娜任艺君武鸿达刘季花
王小静 刘洪娜 任艺君 武鸿达 张 颖 刘季花, 3 李 力, 3
海洋沉积物孔隙水中痕量金属元素的测试分析方法*
王小静1, 2刘洪娜1任艺君1武鸿达1张 颖1, 2刘季花1, 2, 3李 力1, 2, 3①
(1. 自然资源部第一海洋研究所 山东青岛 266061; 2. 自然资源部海洋地质与成矿作用重点实验室 山东青岛 266061; 3. 青岛海洋科学与技术试点国家实验室海洋地质过程与环境功能实验室 山东青岛 266061)
孔隙水是沉积物-海水界面链接沉积物颗粒和上覆水体的一个重要过渡相态, 针对其研究可更好地了解痕量金属在固-液界面的早期成岩过程。近年来, 针对孔隙水中痕量元素研究的方法较为匮乏, 为此建立了一种分析测定海洋沉积物孔隙水中7种痕量金属元素(Mn、Cu、Zn、Ni、Cd、Co、Pb)的方法, 该方法使用Nobias PA1树脂进行富集分离, 再使用电感耦合等离子体质谱(ICP-MS)进行测试, 可针对孔隙水中的痕量金属元素进行准确分析。通过实验结果发现该方法最优实验条件为: Nobias PA1树脂富集时的pH值为5.5~6.0, 洗脱酸浓度为1.3 mol/L硝酸, 体积为1 mL。同时, 样品需进行紫外消解4 h以上以分解有机络合物, 该消解步骤对Cu和Co这两种元素尤其重要。该方法通过加标回收获得Mn、Cu、Ni、Co和Pb的回收率在92%~100%, Zn和Cd的回收率分别为72%和82%; Mn、Cu、Zn、Ni的方法检出限范围为0.03~0.53 nmol/L, Cd、Co、Pb的方法检出限范围为2.66×10-3~8.60×10-3nmol/L, 满足孔隙水中痕量金属浓度的测试需求。同时, 根据检出限计算的结果显示, 孔隙水样品只需1 mL, 即可应用该方法进行测试。应用该方法测试了一根采集于北黄海中部沉积物短柱的孔隙水样品, 测试结果显示其垂相分布合理、较符合早期成岩过程规律。此研究为分析海洋沉积物孔隙水中痕量金属元素提供了一种准确而简便的方法。
小体积孔隙水; Nobias PA1树脂; 痕量金属元素
沉积物-海水界面(sediment-water interface, SWI)是元素地球化学循环和生物系统耦合的主要场所(Sholkovitz, 1989; 吴丰昌等, 1996)。孔隙水作为该界面链接沉积物颗粒和上覆水体的一个重要过渡相态, 针对其研究可更好地了解固液界面生物地球化学循环过程(Elderfield, 1987; Sholkovitz, 1989, 1992; Haley, 2003; Chen, 2015)。在早期成岩过程中, 部分以颗粒态存在的痕量金属(Cu、Pb、Zn等)在SWI发生的一系列生物、物理和化学反应中(包括氧化还原、吸附和解吸、迁移和转化、有机颗粒物降解、生物扰动或者生物灌溉等), 从颗粒物释放至孔隙水, 重新成为溶解态(Santschi, 1990; Aller, 1992; Kalnejais, 2015)。孔隙水和上覆水之间由此形成的浓度梯度差, 可在固液界面产生扩散通量, 痕量金属元素可向上扩散进入水体成为“源”, 进而可能导致水体的二次污染; 也可向下进入沉积物成为“汇”(吴丰昌等, 1996; Abbott, 2015; Dang, 2015; Duan, 2019; Li, 2020)。已有研究报道, 痕量金属元素从沉积物向水体的底层扩散相当于甚至超过河流输入(Kalnejais, 2015), 但针对孔隙水中痕量金属元素的研究较少。
孔隙水通常样品量少(≤10 mL)、含盐分高, 含有机质含量高(0.2~4 mmol/L), 在富有机质的缺氧沉积物中更高(Lukawska-Matuszewska, 2016; Coffin, 2017; Fox, 2018)。目前孔隙水的采集主要是使用孔隙水采样器Rhizon或者离心的方式(Duan, 2019; Li, 2020; Liu, 2021), 痕量金属元素在孔隙水中浓度可能较低(Santos-Echeandia, 2009; Kalnejais, 2015), 且其在采样过程中极易被污染, 无法直接稀释进行准确测定, 因此需要对孔隙水中的痕量金属元素进行预富集分离。目前针对孔隙水的测试方法较少, 但海水较多。针对海水中痕量金属元素的预富集方法包括有机溶剂萃取法(Ellwood, 2008)、共沉淀法(Wu, 1998; Saito, 2006)和固相萃取法(Sohrin, 2008; Milne, 2010)。其中, 螯合树脂富集分离高盐基体内的痕量金属元素, 再使用ICP-MS进行测试是目前国际研究团队最主要使用的测试方法。用于预富集分离的螯合树脂有固定化的8-hydroxyquinoline (8HQ)树脂(Sohrin, 2001)、Toyopearl-650树脂(Hurst, 2008; Milne, 2010)以及Dionex MetPac CC-1树脂(Ho, 2010)等。这些树脂应用广泛, 但还是存在碱金属和碱土金属的亲和性较高(Wang, 2014)且元素富集的pH值范围较窄等问题。近年来, Nobias PA1树脂(Hitachi High-Technology, 日本)被广泛应用于海水中微量元素以及孔隙水中稀土元素的预富集处理(Sohrin, 2008; Biller, 2012; Hatje, 2014; Liu, 2021)。该树脂具有乙二胺四乙酸铵(EDTriA)和亚氨基二乙酸(IDA)官能团, 对海水中的痕量金属元素具有较高的亲和力, 且负载树脂上的痕量金属元素易被硝酸洗脱, 重复利用性高(Sohrin, 2008; Takata, 2011)。同时, 使用该树脂前不需要进行功能团络合, 这显著区别于其他树脂(王美玲等, 2006)。
本研究旨在使用Nobias PA1树脂建立一种专门针对高盐分孔隙水中痕量金属元素(Mn、Cu、Zn、Ni、Cd、Co、Pb)进行测试的分析方法, 以解决孔隙水样品体积小、基质复杂和易污染等问题。本文研究分析了实验pH值、洗脱酸体积和浓度以及紫外消解对实验方法的影响, 报道了该方法的实验空白、检出限和标准海水物质的分析结果。在系统研究的基础上, 确定了最佳实验条件, 成功建立了孔隙水样品中7种痕量金属元素的分析方法, 该分析方法随后被用于测定从北黄海收集的沉积物短柱孔隙水中痕量金属元素的含量。
1 实验方法与样品采集
1.1 仪器和试剂
为避免样品污染和减少试剂空白, 所有实验过程均在自然资源部第一海洋研究所痕量金属超净实验室(千级)的百级洁净工作台中完成。本研究中使用的浓硝酸(HNO3)和浓盐酸(中国化工生产有限公司, 优级纯)均经过酸纯化系统(DST-1000, 美国Savillex公司)的蒸馏过程进行提纯。醋酸铵(NH4Ac)缓冲液由浓醋酸和浓氨水(NH3·H2O) (Optima®, 美国Fisher Scientific公司)制备。采用痕量单元素标准溶液(High-Purity Standards公司)配制了多元素混合标准储备液。各溶液均以Milli-Q水(18.2 MΩ/cm, 美国Millipore公司)配制。
由于孔隙水样品量有限, 紫外消解实验采用2021年8月最新采集的西太平洋中层水(1 000 m; 153.68ºE, 17.80ºN), 其余条件实验采用西太平洋近底的海水(133.67ºE, 9.03ºN)。海水由Niskin瓶采集后, 立即通过AcroPak®过滤器(0.2 μm, Pall公司)收集至预清洗好的LDPE瓶(Nalgene™, Fisher Scientific公司)中, 加入超纯浓HNO3, 酸化至pH值为2。实验过程中使用的离心管(BD FALCON™, Nalgene公司)在10% 盐酸溶液中浸泡24 h后, 用Milli-Q水清洗3~5次, 晾干备用。用于储存缓冲溶液、标准溶液和样品的低密度聚乙烯(LDPE)瓶, 严格按照痕量元素和同位素海洋生物地球化学循环(GEOTRACES)手册建议的清洗流程(Cutter, 2010)。清洗后的LDPE瓶装满pH值为2的Milli-Q水并用聚乙烯袋包装待用。
1.2 仪器参数设置
样品中痕量金属元素采用配备PFA微流量雾化器(美国ESI公司)的电感耦合等离子体质谱(ICP-MS)(iCAP-RQ, 美国Thermo Fisher Scientific公司)进行分析, 不仅可以有效提高仪器的灵敏度, 减少氧化物的干扰, 还可以对微量样品(≥0.5 mL)进行分析。仪器的工作条件见表1。实验分析过程中, 使用含有2% HNO3的标准溶液获得痕量金属元素的标准曲线。为确定仪器空白和校正基线漂移, 每10个样品重复分析1次2% HNO3空白和随机标准溶液。为监测仪器稳定性并避免样品基体干扰, 在样品中加入In内标(10 ng/mL)进行校正。
1.3 实验预富集过程
本研究主要通过两种方式进行孔隙水中痕量金属元素的预富集实验, 即采用手动控制的预富集系统(Biller, 2012)和离线模式的自动预富集系统(seaFAST®, 美国ESI公司)。采用seaFAST的离线模式进行洗脱液体积和紫外消解条件改变的实验, 不仅避免了引进污染的可能性, 而且能准确控制洗脱体积(通常≤1 mL)(Hathorne, 2012; Behrens, 2016)。由于seaFAST比较难实现实验pH值和洗脱酸浓度条件的改变, 因此采用手动控制的预富集系统进行pH值和洗脱酸浓度的影响实验。
表1 本研究中电感耦合质谱(ICP-MS)的工作条件

Tab.1 Operating conditions of the inductively couple mass spectrometry in this study
手动控制的预富集系统采用Biller等(2012)中描述的实验装置, 如图1所示。Nobias-PA1树脂填充后, 经过严格的酸清洗过程(Biller, 2012)。样品预富集前, 先用10 mL 1mol/L HNO3清洗整个富集系统, 然后加入~ 3 mL NH4Ac缓冲溶液调节树脂至实验最优pH条件。紧接着将5 mL孔隙水样品用Milli-Q水(pH=2)稀释至10 mL, 称重并调节好pH后在0.5 mL/min的泵速下通过树脂, 然后在相同的泵速下载入3 mL NH4Ac缓冲溶液去除树脂上残余的Na+, K+, Ca2+和Mg2+(Sohrin, 2008; Wang, 2014)。最后用1mol/L 洗脱硝酸(E-HNO3)(含10 ng/mL In内标)在高纯氮气压力下(3~5 psi)洗脱树脂上富集的痕量金属元素至预称重的15 mL离心管中待测。样品之间, 采用10 mL1 mol/L HNO3清洗整个富集系统。使用后, 管路及树脂浸泡在0.1 mol/L HNO3中。
采用离线模式的seaFAST系统进行条件实验时, 由于10 mL定量环中需要初始样品的体积为12~13 mL,因此我们将6.5 mL孔隙水样品稀释至13 mL。同时使用预富集系统对实验空白和标准海水样品(CASS-6和NASS-7, 加拿大国家研究委员会)进行分析, 以提供质控数据。
1.4 样品的采集
2016年7月在北黄海中部的B20站位(122.74°N, 38.20°E)采集孔隙水样品。用箱式插管采集沉积物短柱后, 立即转移至一个充满N2的手套袋中。将Rhizon采样器(荷兰Rhizonsphere公司)水平插入预先在插管钻的孔中, 并与Teflon管和蠕动泵(LongerPump®Inc., 中国兰格)连接在一起, 形成孔隙水中痕量金属元素的洁净采样系统。在沉积物短柱上的取样间隔0~10 cm为1 cm, 10 cm以下为2 cm。管路中前1 mL孔隙水用于润洗管路, 随后的孔隙水样品收集在15 mL离心管中, 用浓HNO3(Optima®, 美国Fisher Scientific公司)酸化至pH值为2, 并在分析前4 °C冷藏储存。

图1 手动控制的痕量金属元素富集和洗脱装置(修改自Biller et al, 2012)
2 结果与讨论
2.1 紫外消解
在海水中, 痕量金属元素与有机物的络合被认为是主要的络合形式。先前的一项研究表明, 自然存在的有机物会导致痕量金属元素分布中的分异, 因为在富含有机物的沿海水域中, 痕量金属元素与有机物存在强络合作用(Bruland, 1989; Ellwood, 2000)。海水中溶解有机碳(DOC)浓度约为0.04~0.12 mmol/L (Hansell, 1993; Hung, 2007; Walter, 2018), 而孔隙水中的DOC浓度往往高一个数量级, 大多在0.2~4 mmol/L范围内, 有时在富有机质缺氧沉积物中更高(Burdige, 1994; Lukawska-Matuszewska, 2016; Coffin, 2017; Fox, 2018)。如果孔隙水样品中痕量金属元素的有机络合作用强到足以与Nobias PA1树脂有效竞争, 则可能会破坏孔隙水样中痕量金属元素的回收。
本研究中将已加入痕量金属元素混标的样品分成6个13 mL, 将三个平行样品采用紫外消解仪(5144AX-220, 美国Jelight公司)进行紫外消解4 h以上, 以破坏样品中与痕量金属元素络合的强有机配体, 另外3个对照样品则不进行消解。6个样品同时用seaFAST进行富集, 比较消解和未消解的样品中痕量金属元素的浓度(图2)。研究发现未消解的样品中Cu和Co的回收率明显较低, 分别为85.3%±5.02%和62.4%±0.69%。消解以后, Cu和Co的回收率分别达到99.3%±0.17%和98.0%±2.98%。已有研究表明, 紫外消解对于准确定量回收Cu和Co是必须的, 因为这两种金属与有机物之间存在强烈螯合, 以致树脂官能团无法竞争过天然配体(Saito, 2001; Milne, 2010; Biller, 2012)。本研究结果表明, 为准确测定Cu和Co, 需要对孔隙水样品紫外消解。

图2 紫外消解对孔隙水中痕量金属元素回收率的影响
注: 灰色虚线表示回收率为100%
2.2 pH值条件的选择
为了研究pH值对孔隙水中痕量金属元素富集效率的影响, 使用NH4Ac缓冲液调节样品和树脂的pH值在4.00~6.5之间(4.00、4.50、5.00、5.50、5.75、6.00、6.25和6.50), 用于调节样品和树脂pH的缓冲液浓度分别为3.70和0.05 mol/L 。用于实验的海水样品均加入配制的7种痕量金属元素标准混合储备液(最终痕量元素标准液的浓度Cd和Co为2 μg/L, Pb为4 μg/L, Mn、Cu、Zn和Ni为40 μg/L)。对于每个pH值条件下, 实验重复2次。洗脱体积为1 mL, 洗脱液为1.3 mol/L HNO3。
不同pH值条件下痕量金属元素的回收率如图3所示。结果表明, 大多数元素pH值在4.00~6.25范围内, 具有较好的回收率, 均在80%~118%范围内。而Mn在pH<5时, 回收率仅有2%~28%。当pH>6时, 与其他痕量金属元素相比, Co和Zn的回收率明显下降, 在pH=6.5时分别下降至68%和78%。

图3 不同pH值下孔隙水中痕量金属元素的回收率
注: 所有元素的标准偏差<5%; 使用标准混合储备液和背景海水中痕量金属元素浓度加和计算回收率, 灰色虚线表示回收率为100%
pH值对树脂的富集效率具有重要影响, 之前的研究表明, pH值为4.5~8.0时, Nobias PA1树脂对大多数元素均有较好的富集能力(Sohrin, 2008; Takata, 2011; Biller, 2012; Hatje, 2014; Liu, 2021)。对于大部分痕量金属元素, 最优pH值范围为4.2~6.5 (Sohrin, 2008; Takata, 2011), Mn在低pH值下回收率较低, 需保持在pH>5.8才能达到最佳回收(Takata, 2011; Biller, 2012)。本文研究结果显示, Mn在pH=4.0~5.5之间对pH值变化敏感性较高, 因此对于这7种目标元素的最佳pH值范围为5.5~6.0, 回收率为87%~118%。
2.3 洗脱体积
Biller等(2012)和Hatje等(2014)的研究选用0.4~2 mL洗脱液作为最后的洗脱体积, 但也有人认为0.5 mL洗脱液不足以洗脱所有元素(梁杰等, 2017), 因此, 海水样品通常选择>1 mL作为最终洗脱体积。由于孔隙水样品量较少(<10 mL), 较高的富集系数有利于得到更大的富集效率, 因此需要控制样品体积与洗脱体积的比例。小于0.5 mL的洗脱体积不足以用ICP-MS测试所有的痕量金属元素。因此, 选用一系列的洗脱体积(0.5、1、1.5、2 mL), 测试样品回收率来确定最佳洗脱体积。实验条件为pH=5.86, 洗脱酸浓度为1.3 mol/L HNO3。
用seaFAST进行预富集处理, 每个体积进行3次重复, 不同洗脱体积条件下痕量金属元素回收率如图4所示。结果表明, 0.5 mL洗脱液足以洗脱所有的痕量金属元素, 与其他元素相比, Cd在0.5 mL洗脱液中的回收率略低为81%。随着洗脱体积的增加, 所有痕量金属元素的回收率在93%~114%的范围内。0.5 mL洗脱液足以洗脱所有目标元素, 但在ICP-MS测试过程中, 1 mL洗脱液可进行重复测量。因此, 1 mL为最佳洗脱体积。

图4 洗脱液体积对孔隙水中痕量金属元素回收率的影响
注: 所有元素的标准偏差<5%, 灰色虚线表示回收率为100%
2.4 洗脱酸浓度
本实验以HNO3作为洗脱液, 洗脱酸的浓度对元素回收率的影响至关重要。如果浓度过低, 则1 mL不能洗脱所有痕量金属元素; 若酸浓度过高, 测试过程中会对ICP-MS仪器造成损害。本研究中, 分别配制一系列浓度(0.7、1.0、1.3、1.6、2.0、2.2 mol/L)的洗脱液来测试洗脱能力。实验条件为在pH=5.86, 洗脱体积为1 mL, 无紫外消解。
痕量金属元素的回收率随不同洗脱酸浓度的变化如图5所示。当洗脱酸浓度小于1.0 mol/L时, 除Co外, 其余痕量金属元素的回收率均在110%~120%的范围内, 而Co的回收率为82%。当洗脱液浓度增加到1.3~1.6 mol/L时, 大部分痕量金属元素的回收率基本趋于稳定为95%~118%, Cu回收率稍微偏低为89%左右, 继续增加洗脱液浓度回收率显著降低。如上所述, 过高的酸浓度会导致ICP-MS仪器损坏。此外, 之前的研究表明, 过高的酸浓度会导致树脂体积收缩, 降低其寿命(刘瑶等, 2017)。因此, 选择酸浓度为1.3 mol/L为最佳洗脱液浓度。

图5 洗脱酸浓度对孔隙水中痕量金属元素回收率的影响
注: 所有元素的标准偏差<5%; 使用标准混合储备液和背景海水中痕量金属元素浓度加和计算回收率, 灰色虚线表示回收率为100%
2.5 质控数据
根据最佳实验条件的结果, 该方法建立的条件选择为pH=5.5~6.0, 洗脱体积为1 mL, 洗脱酸为1.3 mol/L HNO3, 并进行紫外消解。该实验的方法空白、检测限和回收率如表2所示。本研究中使用Optima优级纯醋酸和氨水以及本实验室二次蒸馏后的硝酸, 通过ICP-MS测试后的本底值均低于检出限, 因此试剂空白可忽略不计。表2中的方法空白主要是在预富集过程中产生的, 检测限是过程空白三倍的标准偏差。结果表明, 方法空白和检测限远低于孔隙水中痕量金属元素的浓度, 该方法适用于孔隙水中痕量金属元素的分析测试。通过在海水中加入一系列混标的方法来确定树脂对痕量金属元素的回收率(Biller, 2012; Hatje, 2014), 大部分金属通过预富集达到的回收率在92%~100%的范围内, Zn和Cd的回收率偏低, 分别为72%和82%。本研究建立的方法对标准海水NASS-7和近岸海水CASS-6进行了测试而分析, 所得结果如表3所示。标准海水的分析结果与标准浓度值相比较, 表现出良好的一致性。
2.6 分析所需的最小孔隙水体积
样品体积的限制是分析孔隙水中痕量金属元素的一个主要影响因素。因此, 有必要确定此分析所需的最小体积。通过对黄渤海21根短柱孔隙水的研究发现, 渤海B67站位(119.94ºN, 37.75ºE)中痕量金属元素的浓度最低(实验室未发表数据)。基于B67站位孔隙水中的最低痕量金属元素浓度, 本文计算了富集1~5 mL孔隙水样品后在1 mL洗脱液中的痕量金属元素浓度(表4)。表4中, 对1 mL样品体积的检出限(LODs)进行了计算, 结果是5 mL样品量时检出限的5倍。通过对比发现, 1 mL孔隙水的样品量足以产生高于该方法的LODs信号值。如果能够获得足够的样品量, 则越高的样品量越有利于获得较高的灵敏度和准确性。
表2 实验方法空白、检出限以及元素回收率

Tab.2 Average procedure blanks, detection limits, and recoveries of all trace metal under optimal experimental conditions
表3 标准海水中(NASS–7、CASS-6)痕量金属元素的分析测试结果(单位: μg/L)
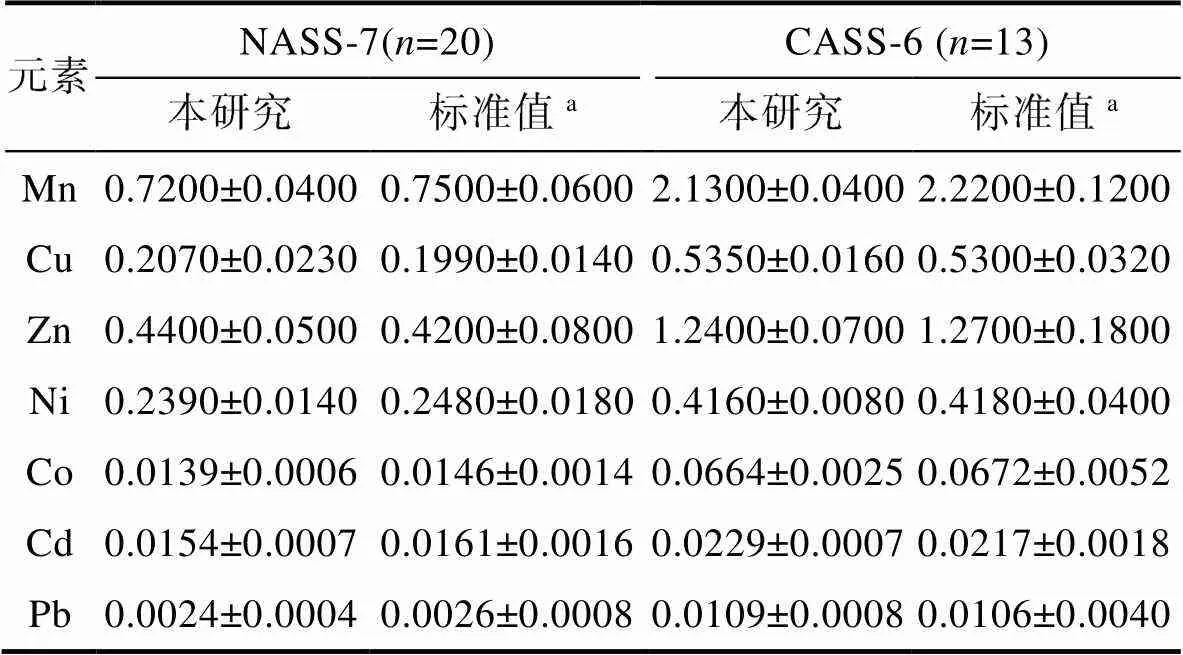
Tab.3 Results of analysis of trace metal concentrations of standard seawater samples (NASS-7, CASS-6) (unit: μg/L)
注:a加拿大国家研究委员会认证的标准海水痕量金属元素值
3 北黄海短柱孔隙水中痕量金属元素的分布
以本研究建立的分析方法, 对北黄海中部B20站位短柱沉积物孔隙水中的痕量金属元素进行了分析, 垂向分布如图6所示。由于近岸孔隙水中Fe、Mn元素含量较高, 采用2% HNO3以1:20稀释的方法直接测试, 其余元素采用本研究方法进行测试。本研究中, Fe、Mn在表层即开始还原, 较高的溶解Mn (28 μmol/L)和Fe (18 μmol/L)出现在孔隙水表层(0~1 cm)。在表层以下, 溶解Mn随着深度的增加逐渐降低然后保持恒定, 而溶解Fe分别在0~4 cm和10~15 cm出现较高的峰值。痕量金属元素在孔隙水表层均有相对较高的含量, 随后随着深度的增加而逐渐降低。溶解Zn、Cd和Pb分别在Fe氧化物的还原释放区(0~4 cm, 10~15 cm)出现峰值, 说明溶解Zn、Cd和Pb的循环过程受Fe氧化物还原释放的影响(Rivera-Duarte, 1994)。此外, Co、Cd和Pb在短柱底部出现了较高的峰值, 这可能与有机质的分解释放有关。总体来讲, 痕量金属元素的垂向分布符合早期成岩过程规律(Froelich, 1979; Canfield, 2009)。
对研究区孔隙水中痕量金属做Pearson相关性分析, 结果如表5所示。统计结果发现Cu、Ni和Co这三种金属之间有显著性正相关(>0.86,<0.01), 但它们与Fe和Mn无显著相关性, 说明孔隙水中这三种元素有相同的来源, 更多可能来自有机质分解释放而不是Fe/Mn氧化物的还原释放(Klinkhammer, 1982; Widerlund, 1996)。此外, Zn与Fe之间有显著正相关(=0.78,<0.01), 说明孔隙水中Zn受到Fe氧化物还原释放的影响。而Cd与Mn之间也存在正相关(=0.49,<0.05), 说明孔隙水中Cd受Mn氧化物还原释放的影响更大。Pb在统计学上与其他元素均无相关性, 说明除了Fe/Mn氧化物还原释放以外可能还受到其他因素的影响。
表4 渤海B67站位孔隙水中最低痕量金属元素的浓度、假设样品体积为1 mL的检测限(LOD)和不同体积的B67站位孔隙水样品(1~5 mL)在1 mL洗脱液中痕量金属元素浓度(单位: nmol/L)

Tab.4 Minimum trace metal concentrations in pore water at B67 station in Bohai sea, the limit of detection (LOD) assuming sample volume of 1mL and estimated trace metal concentrations in different sample volumes (1~5 mL)(unit: nmol/L)

图6 北黄海B20站位孔隙水中痕量金属元素的垂直分布
表5 北黄海B20站位孔隙水中痕量元素的Spearman相关性分析

Tab.5 The spearman correlation among the trace metals in pore water at B20 in the North Yellow Sea
注:**表示在置信度为0.01以内显著性相关;*表示在置信度为0.05以内显著性相关。
4 结论
本研究建立了一种对海洋沉积物孔隙水中7种痕量金属元素(Mn、Cu、Zn、Ni、Cd、Co、Pb)进行分析测定的方法, 该方法应用Nobias PA1树脂进行富集分离, 并用等离子体质谱(ICP-MS)进行了准确测定。通过实验确定了最优实验条件为: Nobias PA1树脂富集时的pH值为5.5~6.0, 洗脱酸浓度为1.3 mol/L HNO3, 体积为1 mL。同时孔隙水样品需进行紫外消解4 h以上来破坏有机物对痕量金属元素的络合, 特别是Cu和Co。本方法对Mn、Cu、Ni、Co和Pb的回收率在92%~100%, Zn和Cd的回收率分别为72%和82%; Mn、Cu、Zn、Ni的检出限范围为0.03~0.53 nmol/L, Cd、Co、Pb的检出限范围为2.66×10–3~8.60×10–3nmol/L, 根据检出限的计算结果, 孔隙水样品仅需1 mL即可满足痕量金属元素浓度的测试需求。应用该方法对北黄海中部沉积物短柱(25 cm)孔隙水中痕量金属元素进行了分析, 结果显示其垂向分布合理、符合早期成岩过程规律。本研究为分析海洋沉积物孔隙水中的痕量金属元素提供了一种准确而简便的方法。
王美玲, ITO M, 2006. NOBIAS chelate-PA1吸附剂固相萃取ICP-AES/ICP-MS测定高盐试样中的金属离子[C] // 2006全国核材料学术交流会论文集. 成都: 中国核学会: 361-366.
刘瑶, 宋金明, 林强, 等, 2017. 离线固相萃取螯合富集分离- ICP-MS测定海水中的稀土元素[J]. 海洋科学, 41(9): 34-40.
吴丰昌, 万国江, 蔡玉蓉, 1996. 沉积物-水界面的生物地球化学作用[J]. 地球科学进展, 11(2): 191-197.
梁杰, 何会军, 麻洪良, 等, 2017. NOBIAS PA1螯合树脂富集——ICP-MS定量测定近海水体中的稀土元素[J]. 海洋科学, 41(10): 58-66.
ABBOTT A N, HALEY B A, MCMANUS J,, 2015. The sedimentary flux of dissolved rare earth elements to the ocean [J]. Geochimica et Cosmochimica Acta, 154: 186-200.
ALLER R C, ALLER J Y, 1992. Meiofauna and solute transport in marine muds [J]. Limnology and Oceanography, 37(5): 1018-1033.
BEHRENS M K, MURATLI J, PRADOUX C,, 2016. Rapid and precise analysis of rare earth elements in small volumes of seawater - method and intercomparison [J]. Marine Chemistry, 186: 110-120.
BILLER D V, BRULAND K W, 2012. Analysis of Mn, Fe, Co, Ni, Cu, Zn, Cd, and Pb in seawater using the Nobias-chelate PA1 resin and magnetic sector inductively coupled plasma mass spectrometry (ICP-MS) [J]. Marine Chemistry, 130-131: 12-20.
BRULAND K W, 1989. Complexation of zinc by natural organic ligands in the central North Pacific [J]. Limnology and Oceanography, 34(2): 269-285.
BURDIGE D J, HOMSTEAD J, 1994. Fluxes of dissolved organic carbon from Chesapeake Bay sediments [J]. Geochimica et Cosmochimica Acta, 58(16): 3407-3424.
CANFIELD D E, THAMDRUP B, 2009. Towards a consistent classification scheme for geochemical environments, or, why we wish the term ‘suboxic’ would go away [J]. Geobiology, 7(4): 385-392.
CHEN J B, ALGEO T J, ZHAO L S,, 2015. Diagenetic uptake of rare earth elements by bioapatite, with an example from Lower Triassic conodonts of South China [J]. Earth-Science Reviews, 149: 181-202.
COFFIN R B, SMITH J P, YOZA B,, 2017. Spatial variation in sediment organic carbon distribution across the Alaskan Beaufort Sea Shelf [J]. Energies, 10(9): 1265.
CUTTER G, ANDERSSON P, CODISPOTI L,, 2010. Sampling and Sample-Handling Protocols for GEOTRACES Cruises [M]. Virginia, USA: GEOTRACES: 41-42. http:// www.geotraces.org/library-88/geotraces-policies/170-sampling-and-sample-handling-protocols-for-geotraces-cruises10.
DANG D H, LENOBLE V, DURRIEU G,, 2015. Seasonal variations of coastal sedimentary trace metals cycling: insight on the effect of manganese and iron (oxy)hydroxides, sulphide and organic matter [J]. Marine Pollution Bulletin, 92(1/2): 113-124.
DUAN L Q, SONG J M, LIANG X M,, 2019. Dynamics and diagenesis of trace metals in sediments of the Changjiang Estuary [J]. Science of the Total Environment, 675: 247-259.
ELDERFIELD H, SHOLKOVITZ E R, 1987. Rare earth elements in the pore waters of reducing nearshore sediments [J]. Earth and Planetary Science Letters, 82(3/4): 280-288.
ELLWOOD M J, 2008. Wintertime trace metal (Zn, Cu, Ni, Cd, Pb and Co) and nutrient distributions in the Subantarctic Zone between 40°–52°S; 155°–160°E [J]. Marine Chemistry, 112(1/2): 107-117.
ELLWOOD M J, VAN DEN BERG C M G, 2000. Zinc speciation in the northeastern Atlantic Ocean [J]. Marine Chemistry, 68(4): 295-306.
FOX C A, ABDULLA H A, BURDIGE D J,, 2018. Composition of dissolved organic matter in pore waters of anoxic marine sediments analyzed by1H nuclear magnetic resonance spectroscopy [J]. Frontiers in Marine Science, 5: 172.
FROELICH P N, KLINKHAMMER G P, BENDER M L,, 1979. Early oxidation of organic matter in pelagic sediments of the eastern equatorial Atlantic: suboxic diagenesis [J]. Geochimica et Cosmochimica Acta, 43(7): 1075-1090.
HALEY B A, KLINKHAMMER G P, 2003. Complete separation of rare earth elements from small volume seawater samples by automated ion chromatography: method development and application to benthic flux [J]. Marine Chemistry, 82(3/4): 197-220.
HANSELL D A, 1993. Results and observations from the measurement of DOC and DON in seawater using a high-temperature catalytic oxidation technique [J]. Marine Chemistry, 41(1/2/3): 195-202.
HATHORNE E C, HALEY B, STICHEL T,, 2012. Online preconcentration ICP-MS analysis of rare earth elements in seawater [J]. Geochemistry, Geophysics, Geosystems, 13(1): Q01020.
HATJE V, BRULAND K W, FLEGAL A R, 2014. Determination of rare earth elements after pre-concentration using NOBIAS-chelate PA-1®resin: method development and application in the San Francisco Bay plume [J]. Marine Chemistry, 160: 34-41.
HO T Y, CHIEN C T, WANG B N,, 2010. Determination of trace metals in seawater by an automated flow injection ion chromatograph pretreatment system with ICPMS [J]. Talanta, 82(4): 1478-1484.
HUNG J J, WANG S M, CHEN Y L, 2007. Biogeochemical controls on distributions and fluxes of dissolved and particulate organic carbon in the Northern South China Sea [J]. Deep Sea Research Part II: Topical Studies in Oceanography, 54(14/15): 1486-1503.
HURST M P, BRULAND K W, 2008. The effects of the San Francisco Bay plume on trace metal and nutrient distributions in the Gulf of the Farallones [J]. Geochimica et Cosmochimica Acta, 72(2): 395-411.
KALNEJAIS L H, MARTIN W R, BOTHNER M H, 2015. Porewater dynamics of silver, lead and copper in coastal sediments and implications for benthic metal fluxes [J]. Science of the Total Environment, 517: 178-194.
KLINKHAMMER G, HEGGIE D T, GRAHAM D W, 1982. Metal diagenesis in oxic marine sediments [J]. Earth and Planetary Science Letters, 61(2): 211-219.
LI L, ZHEN X T, WANG X J,, 2020. Benthic trace metal fluxes in a heavily contaminated bay in China: does the sediment become a source of metals to the water column? [J]. Environmental Pollution, 257: 113494.
LIU H N, LI L, WANG X J,, 2021. Determination of rare earth elements in pore water samples of marine sediments using an offline preconcentration method [J]. Archives of Environmental Contamination and Toxicology, 81(4): 553-563.
LUKAWSKA-MATUSZEWSKA K, 2016. Contribution of non-carbonate inorganic and organic alkalinity to total measured alkalinity in pore waters in marine sediments (Gulf of Gdansk, S-E Baltic Sea) [J]. Marine Chemistry, 186: 211-220.
MILNE A, LANDING W, BIZIMIS M,, 2010. Determination of Mn, Fe, Co, Ni, Cu, Zn, Cd and Pb in seawater using high resolution magnetic sector inductively coupled mass spectrometry (HR-ICP-MS) [J]. Analytica Chimica Acta, 665(2): 200-207.
RIVERA-DUARTE I, FLEGAL A R, 1994. Benthic lead fluxes in San Francisco Bay, California, USA [J]. Geochimica et Cosmochimica Acta, 58(15): 3307-3313.
SAITO M A, MOFFETT J W, 2001. Complexation of cobalt by natural organic ligands in the Sargasso Sea as determined by a new high-sensitivity electrochemical cobalt speciation method suitable for open ocean work [J]. Marine Chemistry, 75(1/2): 49-68.
SAITO M A, SCHNEIDER D L, 2006. Examination of precipitation chemistry and improvements in precision using the Mg(OH)2preconcentration inductively coupled plasma mass spectrometry (ICP-MS) method for high-throughput analysis of open-ocean Fe and Mn in seawater [J]. Analytica Chimica Acta, 565(2): 222-233.
SANTOS-ECHEANDIA J, PREGO R, COBELO-GARCÍA A,, 2009. Porewater geochemistry in a Galician Ria (NW Iberian Peninsula): implications for benthic fluxes of dissolved trace elements (Co, Cu, Ni, Pb, V, Zn) [J]. Marine Chemistry, 117(1/2/3/4): 77-87.
SANTSCHI P, HÖHENER P, BENOIT G,, 1990. Chemical processes at the sediment-water interface [J]. Marine Chemistry, 30: 269-315.
SHOLKOVITZ E R, 1989. Artifacts associated with the chemical leaching of sediments for rare-earth elements [J]. Chemical Geology, 77(1): 47-51.
SHOLKOVITZ E R, 1992. Chemical evolution of rare earth elements: fractionation between colloidal and solution phases of filtered river water [J]. Earth and Planetary Science Letters, 114(1): 77-84.
SOHRIN Y, KINUGASA M, OKAMURA K,, 2001. Determination of trace metals in the ocean by MAF-8HQ column extraction-ICP-MS [J]. Analytical Sciences, 17(S1): 1-4.
SOHRIN Y, URUSHIHARA S, NAKATSUKA S,, 2008. Multielemental determination of GEOTRACES key trace metals in seawater by ICPMS after preconcentration using an ethylenediaminetriacetic acid chelating resin [J]. Analytical Chemistry, 80(16): 6267-6273.
TAKATA H, AONO T, TAGAMI K,, 2011. Determination of naturally occurring uranium concentrations in seawater, sediment, and marine organisms in Japanese estuarine areas [J]. Journal of Radioanalytical and Nuclear Chemistry, 287(3): 795-799.
WALTER S R S, JAEKEL U, OSTERHOLZ H,, 2018. Microbial decomposition of marine dissolved organic matter in cool oceanic crust [J]. Nature Geoscience, 11(5): 334-339.
WANG B S, LEE C P, HO T Y, 2014. Trace metal determination in natural waters by automated solid phase extraction system and ICP-MS: the influence of low level Mg and Ca [J]. Talanta, 128: 337-344.
WIDERLUND A, 1996. Early diagenetic remobilization of copper in near-shore marine sediments: a quantitative pore-water model [J]. Marine Chemistry, 54(1/2): 41-53.
WU J F, BOYLE E A, 1998. Determination of iron in seawater by high-resolution isotope dilution inductively coupled plasma mass spectrometry after Mg(OH)2coprecipitation [J]. Analytica Chimica Acta, 367(1/2/3): 183-191.
DETERMINATION OF TRACE METALS IN POREWATER SAMPLES OF MARINE SEDIMENTS
WANG Xiao-Jing1, 2, LIU Hong-Na1, REN Yi-Jun1, WU Hong-Da1, ZHANG Ying1, 2, LIU Ji-Hua1, 2, 3, LI Li1, 2, 3
(1. First Institute of Oceanography, Ministry of Natural Resources, Qingdao 266061, China; 2. Key Laboratory of Marine Geology and Metallogeny, Ministry of Natural Resources, Qingdao 266061, China; 3. Laboratory for Marine Geology, Pilot National Laboratory for Marine Science and Technology (Qingdao), Qingdao 266061, China)
Porewater is an important transitional phase between sediment particles and overlying water at the sediment-seawater interface, and the study of porewater can provide a better understanding of the early diagenetic process of trace metals. In recent years, the methods for trace metals in pore water are relatively scarce. Therefore, a highly sensitive and precise method for the determination of 7 trace metals (Mn, Cu, Zn, Ni, Cd, Co, Pb) in porewater of marine sediment was established. The method uses Nobias PA1 resin preconcentrated trace metals in porewater samples, and then the samples are analyzed by inductively coupled plasma mass spectrometry (ICP-MS). The experiment results show that the optimal experimental conditions could be reached when pH in the preconcentration step was 5.5~6.0, the elution solution volume was 1 mL, and the elution acid concentration was 1.3 mol/L HNO3. In addition, the samples shall be ultraviolet digested for more than 4 h to destroy organic complexes, which procedure is especially important for Cu and Co. The recoveries of Mn, Cu, Ni, Co, and Pb were 92%~100%, whilst those of Zn and Cd were 72% and 82%, respectively. The detection limits were also reported; for Mn, Cu, Zn, and Ni, it was 0.03~0.53 nmo/L, whilst for Cd, Co, and Pb, it was 2.66×10-3~8.60×10-3nmol/L, which satisfies the needs for porewater analysis, and only 1 mL of porewater sample is required for the measurement and calculation of dissolved trace metals. This method was applied to test a porewater sample collected from a short sediment column in the middle of the North Yellow Sea. The test results show that its vertical distribution is reasonable and more in line with the law of early diagenesis. This method provides an accurate and simple way to analyze the seven trace metals in marine porewater samples in a minimum sample volume of 1 mL.
small volume pore water; Nobias PA1 resin; trace elements
P736
10.11693/hyhz20220100010
*国家自然科学基金, 41776095号; 国家重点基础研究发展计划, 2013CB429704号。王小静, 助理工程师, E-mail: wxj1130@ fio.org.cn
李 力, 研究员, E-mail: Li.Li@fio.org.cn
2022-01-13,
2022-04-11
