内耳不完全分隔Ⅲ型畸形致复发性脑膜炎1例*
2022-09-20龙丹张春林
龙丹 张春林
1 遵义医科大学附属医院耳鼻咽喉头颈外科(遵义 563000)
先天性内耳畸形是指胚胎期由内耳发育障碍引起的内耳结构异常,可伴自发性脑脊液漏,其原因是蛛网膜下腔直通畸形的内耳,脑脊液长期冲击内耳的薄弱处使之破裂形成漏口,从而脑脊液进入中耳,中耳腔的致病菌可以逆行进入颅内导致脑膜炎反复发作。内耳畸形导致的自发性脑脊液漏的治疗根本在于修补内耳漏口,常用的手术方式有多种,需结合患者病史、临床症状及畸形特点进行个体化选择。耳蜗不完全分隔(incomplete partition type, IP)Ⅲ 型是内耳畸形中一种极少见的分型,临床上缺乏其伴自发性脑脊液漏的相关报道。遵义医科大学附属医院耳鼻咽喉头颈外科收治1例IP-Ⅲ 型伴自发性脑脊液耳鼻漏并致脑膜炎反复发作的病例,手术治疗后临床效果满意,现报道如下。
1 病例资料
21岁男性患者,因自幼右耳听力下降伴鼻腔流液,反复头痛、发热17年入院。患者自幼发现右耳听力下降,伴鼻腔间断性流出清亮液体,未行进一步诊疗。17年前出现头痛、发热,就诊于当地医院,诊断为“颅内感染”,予以抗感染治疗后恢复。此后患者头痛、发热症状反复发作,发作时伴有意识障碍、小便失禁等,多次就诊于当地医院,诊断为“脑膜炎”,抗炎治疗后好转。1年前于外院行脑脊液耳漏修补术(具体不详),术后6月患者再发鼻腔流液、头痛、发热等症状,当地医院诊断为“脑脊液漏复发,脑膜炎”,抗炎治疗后控制。为求进一步治疗,患者于2019年9月19日就诊于遵义医科大学附属医院耳鼻咽喉头颈外科,专科检查:耳廓无畸形,右耳后见长约5 cm陈旧性疤痕,乳突区无红肿、压痛,右外耳道闭锁,鼓膜未窥及。纯音听阈测试示:右耳重度感音神经性聋(全聋型)。鼻内镜检查可见右侧咽鼓管咽口有清亮液体流出,按压右侧颈部时明显增多。颞骨CT:右侧鼓室、乳突及岩尖软组织影;耳蜗底回与内听道底的骨质间隔部分缺失,蜗孔扩大,蜗轴缺失;内听道底异常膨大,与耳蜗底回融为一大腔(图1)。入院诊断:①右侧先天性内耳畸形(IP-Ⅲ 型);②右侧脑脊液耳鼻漏;③右耳全聋;④右外耳道封闭术后。
患者在全麻下行右侧岩骨次全切术进行脑脊液漏的修补。手术步骤:沿原耳后瘢痕处做切口,离断外耳道皮肤,探查原耳道口封闭可,取肌骨膜瓣覆盖耳道口内侧,加固已封闭的外耳道口。探查见乳突骨质部分缺如,乳突腔内充满瘢痕组织、脂肪组织及骨蜡,予以清理,切除颞骨的残余气房,探查镫骨完整,前庭窗及圆窗无漏口;轮廓化中颅窝脑板及乙状窦及后颅窝脑板,探查岩尖见骨质缺损,填充大量骨蜡,予以清理,未见漏口。轮廓化迷路下方后颅窝脑板,见后颅窝骨板部分缺如,硬脑膜暴露,见耳蜗导水管内侧一漏口,持续脑脊液漏出(图2),制备条形颞肌组织塞入漏口,见脑脊液流出停止。向前轮廓化咽鼓管口周围气房,取骨蜡及肌肉组织填塞封堵咽鼓管口,取腹部脂肪填塞术腔,再取带蒂颞肌瓣,向下翻转覆盖术腔。分层缝合皮肤,颞部皮下留置负压引流,加压包扎。术后给予抗生素预防感染及对症支持治疗,按时换药,伤口愈合良好,无脑脊液漏的征象,出院并按时随访。随访15个月,患者无鼻腔流液及脑膜炎发作。
2 讨论
内耳畸形是先天性感音神经性聋的重要原因之一,发病率约为1/2 000~1/6 000[1],其中IP-Ⅲ 畸形更为少见,发病率约占内耳畸形的3.3%[2]。IP-Ⅲ 畸形与POU3F4基因突变所致的X连锁遗传性聋有关[3],常见男性发病,女性携带。本例男性患者自幼单侧听力下降,由于对侧听力正常,患侧听力下降未被家长发现而长期漏诊;此外,患者反复发作脑膜炎以治疗脑膜炎为主,从而忽略了病因诊断。IP-Ⅲ 畸形的诊断主要依靠颞骨CT检查,其特征性表现为耳蜗外部轮廓与正常耳蜗相同,蜗轴缺失,耳蜗底回与膨大内听道底相通[4]。临床上对复发性脑膜炎合并听力下降者,应考虑内耳畸形伴自发性脑脊液漏的可能,需行颞骨CT及MRI检查以明确诊断[5]。

图1 术前颞骨CT及头颅MRI a. 白色箭头示:耳蜗底回与内听道底的骨质间隔部分缺失,蜗孔扩大,蜗轴缺失;内听道底异常膨大,与耳蜗底回融为一大腔; b. 黑色箭头示:右侧耳蜗导水管扩大; c. 黄色箭头示:耳蜗底回、中回、顶回,内听道底异常膨大,与耳蜗底回相通; d. 右侧颞骨见片状长T2信号
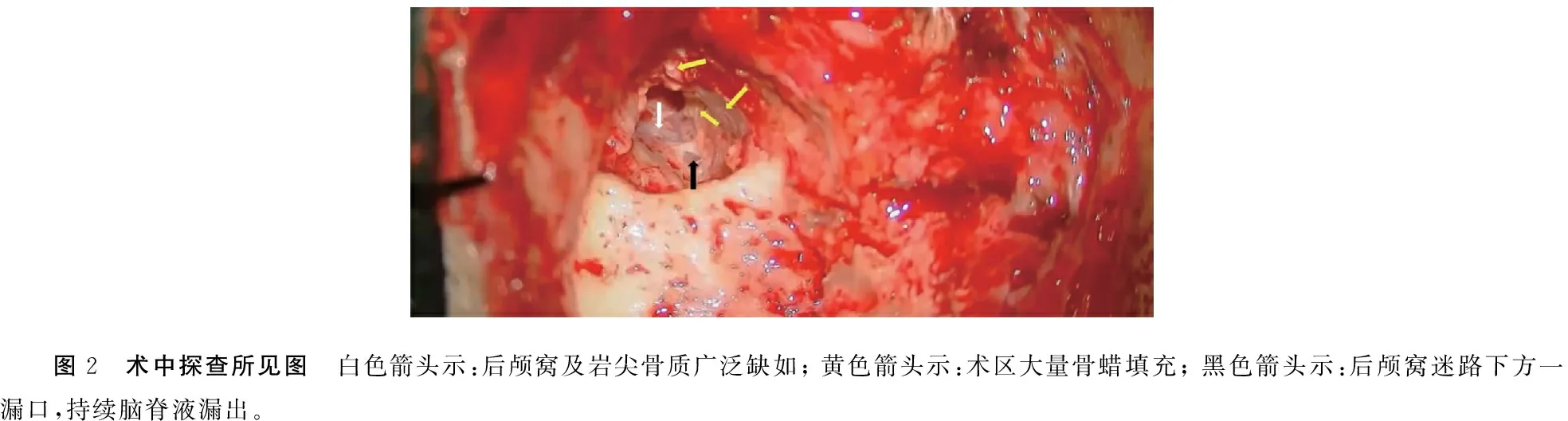
图2 术中探查所见图 白色箭头示:后颅窝及岩尖骨质广泛缺如; 黄色箭头示:术区大量骨蜡填充; 黑色箭头示:后颅窝迷路下方一漏口,持续脑脊液漏出。
内耳畸形是导致自发性脑脊液漏的常见原因之一,其原因为蛛网膜下腔与内耳之间存在异常通道,导致这一现象的原因未明,其中先天性骨质缺损和蛛网膜颗粒异常是被普遍认可的两种理论[6],脑脊液可通过发育异常的骨质缺损处或脑脊液长期冲击内耳的薄弱处产生漏口,而最常见的漏口部位为前庭窗及圆窗[7]。临床上IP-Ⅲ 畸形伴自发性脑脊液漏的病例十分罕见,李万鑫等[8]和卢宇涵等[5]均报道了几十例行人工耳蜗植入的内耳畸形病例,术前皆无自发性脑脊液漏及脑膜炎发作的病史。Wang等[9]报道了18例内耳畸形伴脑脊液漏的病例,多为IP-Ⅰ和IP-Ⅱ畸形,无IP-Ⅲ 畸形病例。其原因可能是IP-Ⅲ 型患者的镫骨足板结构相对正常[4],相对于其他类型的内耳畸形,不易在薄弱的前庭窗处形成漏口。
先天性内耳畸形伴脑脊液耳鼻漏治疗的根本在于手术修补内耳漏口,并有效填塞,预防其再发。手术方式[9~11]包括经耳道鼓室径路、经乳突径路、经颅中窝径路和经迷路径路等,需结合患者病史、临床特点、听力状况等进行个体化的选择。国内报道前两种径路常见,但存在一定手术失败率从而导致脑脊液漏复发[7,9,11],Wang等[9]对18例内耳畸形伴脑脊液漏的患者进行手术,3例经耳道鼓室径路、15例经乳突径路手术治疗,经鼓室径路者有1例术后6月复发。Deng等[11]报道了13例小儿内耳畸形伴脑脊液漏的患者经耳道鼓室径路手术,有1例术后9天复发。而临床上常采用岩骨次全切除术对复杂性脑脊液漏进行修补[12~14],岩骨次全切除能有效地对抗持续存在的脑脊液冲击,从而降低原漏口及其他薄弱处再发脑脊液漏。Kronenberg等[13]和Magliulo等[14]均对脑脊液漏患者行岩骨次全切除术,其中大多患者术前至少有1次耳内入路脑脊液漏修补失败的病史,最终均修补成功。本例患者1年前于外院诊断为脑脊液耳漏后行手术修补,术后6月再发脑脊液漏及脑膜炎,失败的主要原因可能是术中未明确漏口位置,未达到有效修补;此外,填塞术腔时过多使用骨蜡,骨蜡与自体组织难以融合,远期易与自体组织形成潜在间隙,从而再发脑脊液漏。考虑本例患者内耳畸形特点,漏口隐匿,单耳发病,一次脑脊液漏修补失败史等,故选用岩骨次全切除术,术中探查发现后颅窝脑膜暴露,后颅窝迷路下方见漏口,脑脊液持续漏出,采用颞肌、骨蜡及脂肪等行脑脊液漏修补术;脑脊液漏口可用肌肉、颞肌筋膜多层填塞,最后使用腹部脂肪以及生物胶等封闭术腔。目前脑脊液漏修补术中多层填塞理念得到广泛认可,可显著降低术后脑脊液漏复发率[9,15]。该患者术后随访15月,未再发生脑脊液漏及脑膜炎,提示由内耳畸形引起的复杂性脑脊液漏,岩骨次全切除术可达到良好的治疗效果。
