动植物油脂加氢脱氧贵金属系催化剂的研究进展
2022-09-19龚绍峰龚建议雷稳强肖新生
龚绍峰,龚建议,雷稳强,肖新生
(1.湖南科技学院 化学与生物工程学院,湖南 永州 425199; 2.中国石化催化剂有限公司,北京 100029)
随着经济的发展,人们对燃料和化工品的需求不断增大,而传统的化石资源生产路线带来的环境问题,引发了世界范围内人们对可再生资源开发利用的关注,尤其是将可再生的生物质资源转化为绿色液态燃料或高附加值的化工产品[1-4]。在诸多可再生资源中,动植物油脂具有能量密度高、结构类似于石油基燃料、加工工艺路线简单等优势,将其转化为液体燃料(如柴油、航空煤油、润滑油)和化学品(如α-烯烃、脂肪酸、脂肪醇等),无论从技术上还是经济上有着比其他种类生物质更佳的可行性[3, 5- 6]。迄今为止,动植物油脂转化路线主要有3种:酯交换反应转化为脂肪酸甲酯,裂化反应转化为短链烃类,加氢脱氧转化为长链烃类(见图1)[4]。其中在生物基燃料升级的背景下,加氢脱氧路线近年来受到广泛的关注。通过加氢脱氧,动植物油脂可以完全转化为可以替代化石资源的碳氢化合物,相对于其他2种路线,加氢脱氧路线由于经济性更高、更加绿色环保以及可以利用现有的石化加氢装置等优点,具有更广阔的应用前景[3- 4, 7]。围绕着动植物油脂的加氢脱氧,科研工作者进行了大量的研究,分别开发了金属硫化物催化剂、贵金属催化剂、镍催化剂和金属磷化物/氮化物催化剂等。其中贵金属催化剂由于其高活性、水热稳定性优异、兼具脱氧和异构双功能以及无污染物产生等优点,在动植物油脂加氢脱氧处理领域有着广阔的应用前景。

图1 甘油三酯类化合物主要转化路线
近年来,科研工作者围绕动植物油脂加氢脱氧进行了一些研究[7-9],但大部分主要集中在原料、反应条件、过渡金属硫化物催化剂等方面,对于贵金属系催化剂及其反应机制、催化剂失活的研究不够深入。由于天然的动植物油脂成分复杂,许多研究工作者以脂肪酸如棕榈酸(十六烷酸)、硬脂酸(十八烷酸)和油酸(顺-9-十八碳烯酸)及其酯类作为模型化合物,研究了不同催化剂催化加氢脱氧性能。模型化合物的研究可以排除许多干扰因素,对比较各类催化剂的催化性能、理解反应动力学和反应机制方面都具有十分重要的意义。另一方面,动植物油脂加氢脱氧后的产物主要为C15~C18的直链正构烷烃,这些直链烷烃的低温流动性能较差,往往需要进一步异构化来改善其低温流动性能,一个高效的方法是采用同一个催化剂一步实现加氢脱氧和异构双重功能。因此,本文对近年来采用贵金属催化剂催化动植物油脂及相关模型化合物加氢脱氧,一步法实现加氢脱氧和异构双重功能及催化剂失活等方面的相关研究进展进行综述,以供参考。
1 加氢脱氧路线及氢耗
科研工作者们[2, 10-11]在不同的催化剂体系上研究了动植物油脂的加氢脱氧机制。普遍认为有3种脱氧路线(见图2):①脱羰反应(Decarbonylation,DCO),消耗2 mol H2,生成烷烃链上减少一个碳原子的烷烃、H2O和CO;②脱羧反应(Decarboxylation,DCO2):消耗1 mol H2,生成烷烃链上减少一个碳原子的烷烃和CO2;③加氢脱氧反应(HDO):消耗4 mol H2,生成烷烃链上碳原子数不变的烷烃和H2O。其中DCO和DCO2反应生成的产物CO、CO2和H2O可能会进一步发生水煤气变换(方程1)及甲烷化(方程2和3)等副反应[12]。不同的反应机制对液体产物的收率及氢耗影响很大,从而影响该反应的经济性。从上述反应机制可以发现,HDO路线的液体产物质量收率最高(烷烃链上碳原子数不变);虽然DCO和DCO2路线的氢耗低于HDO路线,但是如果生成CO和CO2,进一步发生甲烷化副反应(CO转化为CH4消耗3 mol H2;CO2转化为CH4消耗4 mol H2),DCO和DCO2路线的氢耗都达到5 mol,反而比HDO路线的4 mol更高,因此HDO路线不论在氢耗还是液体产物收率上都比DCO和DCO2路线更加经济。

图2 甘油三酯类化合物加氢脱氧路线
CO2+H2=CO+H2O
(1)
CO2+4H2=CH4+2H2O
(2)
CO+3H2=CH4+H2O
(3)
2 动植物油脂加氢脱氧贵金属系催化剂
2.1 加氢脱氧机制
负载型贵金属催化剂与催化性能之间的构效关系近年来得到了广泛的研究,普遍认为催化剂的高加氢活性与其氢解离能力和氢溢出效应相关:金属活性位点活化氢气,而载体和金属-载体界面则将活化的氢转移到反应底物[10]。负载型贵金属催化剂上脂肪酸的加氢脱氧也有类似的金属和载体之间的协同效应(见图3)[11-13]。在脂肪酸的加氢脱氧过程中,氢气解离吸附在金属颗粒表面形成活化氢,而含氧底物则被吸附在金属位点或载体的氧空位上并活化(极性分子,例如含有羰基的分子,容易通过含氧官能团与载体之间发生相互作用[11,14])。随后,解离的氢从金属活性位点转移到被吸附且活化的含氧底物上,导致C—O的断裂并伴随着水的形成。
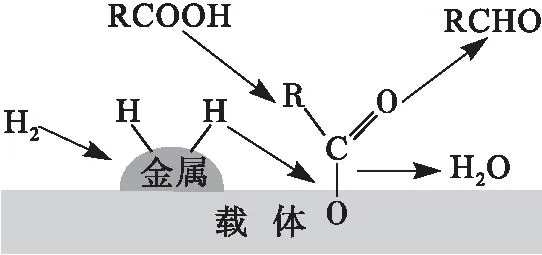
图3 负载型贵金属催化剂上加氢脱氧机制
Chen等[15]研究了Pt/SAPO-11催化剂上油酸甲酯的加氢转化并提出了可能的反应机制(见图4)。首先是不饱和的油酸甲酯快速加氢生成饱和的硬脂酸甲酯。然后,硬脂酸甲酯在Pt活性位点上发生氢解反应生成硬脂酸。接下来,硬脂酸通过3种途径进行脱氧:HDO路径生成的十八醇,DCO路径生成的十七醇,以及DCO2路径生成正十七烷。HDO路径生成的十八醇和DCO路径生成的十七醇进一步加氢分别生成正十八烷和正十七烷。最后,正十七烷和正十八烷发生异构化反应生成带支链的异构烷烃。Bie等[13]在研究Rh/ZrO2催化剂上棕榈酸甲酯的加氢脱氧时也提出了类似的反应机制。不过Bie等[13]认为棕榈酸加氢过程中先生成十六醛中间体,十六醛再经过连续加氢和脱羰基分别生成正十六烷和正十五烷。
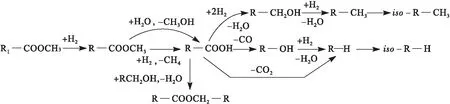
注:R1.C17H33;R.C17H35图4 Pt/SAPO-11催化剂上油酸甲酯加氢转化反应机制
2.2 单活性金属
Pt、Pd、Ru、Rh等负载型贵金属催化剂在多种反应中都有着广泛的应用,其中最重要的是加氢反应,包括含氧化合物的选择性加氢和碳氢化合物的异构化、环化、加氢裂化等。负载型贵金属催化剂由于具有较高的H2活化能力,且不易因水失活,在动植物油脂加氢处理领域引起了广泛的研究兴趣。早期,研究者在无氢或低氢压条件下,研究了一系列金属(Pd、Pt、Ru、Ir、Rh、Os、Ni 和 NiMo)和载体(活性炭、SiO2、Al2O3、MgO 和 Cr2O3)的脂肪酸脱氧性能[16-22],结果发现不同金属的脱氧活性依次为Pd>Pt>Ni>Rh>Ir>Ru>Os,这些催化剂上的脱氧路线主要是 DCO 和 DCO2,生成比原始脂肪酸少一个碳原子数的碳氢化合物。活性炭负载的Pd/C和Pt/C催化剂显示出了极高的硬脂酸脱氧活性和十七烷选择性,但两种催化剂上的反应路线不同,Pd/C催化剂以DCO2为主,而Pt/C催化剂以DCO为主。
近年来,Lopez-Ruiz等[23]研究了无氢条件下碳负载Pt纳米颗粒催化剂上庚酸的加氢脱氧反应,结果发现在液相和气相操作条件下,主要反应路线都为DCO路线。在气相条件下,可得到高选择性的α-烯烃产物,但是升高反应压力α-烯烃会异构化生成内烯烃;在液相条件下,产物则主要是内烯烃,这主要是由于液相反应中脱羰生成α-烯烃传质较慢,进一步发生双键异构生成了内烯烃。反应后催化剂上Pt的平均粒径从1.9 nm增大到2.3 nm,说明反应过程中发生Pt烧结现象,但并不严重。虽然对废催化剂进行了再生研究,但未能恢复催化剂的活性。Ahmadi等[24]研究了不同载体(分子筛SAPO-34、DNL-6、RHO和水滑石)负载的Pt催化剂上油酸的加氢脱氧,结果发现各催化剂都显示了较高的催化脱氧活性,主要脱氧路线都是DCO2路线。其中Pt/SAPO-34催化剂显示出了最高的十七烷和十二烷基苯选择性,作者认为主要归结于SAPO-34的强酸性和小孔径,大量的强酸中心有利于脱氧反应发生,而小孔径则限制了脱羧生成的直链烷烃进一步发生异构化反应。同时,发现升高反应温度可以进一步提高DCO2反应的选择性。以硝酸四氨合铂(Ⅱ)为前驱体制备的催化剂具有最好的Pt分散性和脱氧活性。Kon等[25]研究了不同载体(Nb2O5、ZrO2、CeO2、MgO、Al2O3、SiO2、TiO2、BEA和MFI)负载的Pt催化剂上脂肪酸和甘油三酯的加氢脱氧,结果发现Pt/Nb2O5催化剂具有最高的催化加氢脱氧活性,在压力0.8 MPa和温度180~250℃下可以实现脂肪酸和甘油三酯的有效加氢脱氧,得到收率高达88%~100%的与原料碳原子数相同的直链烷烃。原位红外光谱研究发现,Nb阳离子与羰基氧之间存在Lewis酸碱相互作用,作者认为Nb2O5载体与活性金属Pt之间产生的强相互作用导致了其高的催化加氢脱氧活性。该研究发现,Pt/Nb2O5催化剂上的脱氧路线几乎都是HDO路线,而其他催化剂上的脱氧路线则以DCO/DCO2路线为主,但作者并未详细讨论载体对脱氧路线的影响。Shao等[26]采用溶胶-凝胶法制备了系列Nb2O5-SiO2载体,研究了Pd/Nb2O5-SiO2催化剂上棕榈酸和甘油三酯的加氢脱氧,结果发现Nb2O5的添加有利于促进C—O断裂和抑制C—C断裂,从而大幅提高催化剂的催化加氢脱氧活性和HDO脱氧路线的选择性。4%Pd/10%Nb2O5-SiO2催化剂具有良好的催化脱氧活性和稳定性,在170℃和2.5 MPa反应条件下可得到收率高达94%的直链烷烃,连续运行150 h未发现催化剂失活。Zhou等[27]研究了Pt/Al2O3和Rh/Al2O3催化剂上微藻油的加氢脱氧,结果发现两种催化剂上加氢脱氧路线几乎不受温度、压力和氢油比等反应条件影响,主要为DCO/DCO2路线。两种催化剂,尤其是Rh/Al2O3催化剂上发生了明显的甲烷化反应,生成了大量的CH4。Pt/Al2O3催化剂比Rh/Al2O3具有更高的催化脱氧活性,在310℃和3.45 MPa时,烃类产物收率可达到76.5%。He等[28]研究了一系列负载型Ru催化剂上脂肪酸酯的加氢脱氧,结果发现Ru/TiO2催化剂在相对温和条件下(200℃,3 MPa)实现了硬脂酸乙酯的高效脱氧,并且十七烷和十八烷的选择性高达99.9%。相同条件下,Ru/SiO2、Ru/Al2O3和Pd/TiO2等催化剂的活性较低(转化率<6%),作者把Ru/TiO2的高活性归结于Ru和TiO2之间的协同效应。脂肪酸酯的酯基能吸附在TiO2表面,然后与Ru纳米颗粒上的解离氢发生反应实现脱氧。Ali等[29]研究了水相中负载型Ru催化剂上微藻油的加氢脱氧,结果发现以高亲水性介孔碳材料为载体的Ru/C催化剂在较低温度(140℃)下也能实现微藻油的高效脱氧,催化剂循环使用8次未发现明显失活,具有良好的活性稳定性。然而反应路线以DCO为主,产物主要为正十七烷。Bie等[13]采用间歇式反应器研究了Rh/ZrO2催化剂上棕榈酸甲酯的加氢脱氧,结果发现在温度270℃、H2压力8 MPa、时间60 min条件下可以实现棕榈酸甲酯的高效脱氧,反应路线以DCO/DCO2为主,产物主要为正十五烷。Mao等[30]采用催化转移加氢和水相重整技术研究了不同溶剂体系里Pd/C催化剂上三油酸甘油酯的水热加氢脱氧,结果发现不同溶剂体系下,反应路线都以DCO/DCO2为主。以90%十氢萘+10%水为溶剂时,类柴油烷烃(C17+C18)收率高达71.91%。溶剂的种类对产物分布有着很大的影响,可能主要是因为溶剂生成的原位氢比水相重整生成的原位氢更具活性。从上述研究可以发现:一方面Pt/Nb2O5[25]、Pd/Nb2O5-SiO2[26]、Ru/TiO2[28]和Ru/C[29]等催化剂显示了极高的低温脱氧活性,然而这些催化剂上的贵金属含量仍然较高,高昂的催化剂成本对其工业化应用会产生极大的限制。因此,接下来需要进一步提高催化剂的活性和稳定性并降低催化剂上贵金属的含量。另一方面载体和贵金属种类以及载体与活性金属之间的搭配对脱氧路线都有着极大的影响。因此,在催化剂的性质和结构对反应机制、反应路径的影响方面需要进一步深入研究。
2.3 多活性金属
金属的特性往往会因为加入其他金属形成合金而改变,从而对化学吸附的强度,催化剂反应活性、稳定性和选择性等产生影响[31]。通过在贵金属中加入其他廉价的活性金属产生协同效应一方面可以进一步提高催化剂的活性和稳定性,另一方面可以降低贵金属的含量,从而降低催化剂的成本。Bhattacharjee等[32]研究了介孔泡沫硅(MCF)负载Fe-Pd-Ni三金属催化剂上油酸的加氢脱氧,结果发现提高CO2分压和采用MCF为载体都有助于提高HDO反应路线的选择性。在优化条件(反应温度278℃,反应时间4 h,CO2分压2 MPa,H2分压4 MPa)下,采用Fe-Pd-Ni/MCF催化剂,十八烷收率可达93%。作者认为这主要归结于两个方面:一是MCF载体的三维笼状结构和较大的孔隙率可以提高金属分散度和减少反应的扩散阻力;二是Fe纳米颗粒表面形成的Pd-Ni合金吸附并解离H2,然后H溢流到Fe纳米颗粒上提高催化剂的活性以及HDO路线的选择性。Domínguez-Barroso等[33]在无外加氢气和亚临界水条件下,以Pt-Ni/Al2O3和Pd/C为组合催化剂,研究了葵花籽油的加氢脱氧,结果发现首先是甘油三酯水解生成脂肪酸和甘油,然后甘油发生水相重整生成H2和CO2,最后脂肪酸与生成的H2发生加氢脱氧生成类柴油烃类产物。在亚临界水条件下,Pt-Ni/Al2O3催化剂促进了甘油的重整和脂肪酸的加氢脱氧,Pt-Ni/Al2O3和Pd/C催化剂的组合使得脂肪酸进一步脱羧形成C17烷烃。Kon等[34]制备了多种贵金属(Pt、Pd、Rh、Ru)和MoOx共负载在TiO2载体上的催化剂,并研究了其催化加氢脱氧性能,结果发现PtMoOx/TiO2催化剂显示出最高的催化加氢脱氧活性,并且其脱氧路线完全为HDO路线。原位红外光谱研究表明,PtMoOx/TiO2催化剂的高活性归结于Pt与MoOx/TiO2载体上的Lewis酸中心的协同作用。Zharova 等[35]研究了PtSn/Al2O3催化剂上菜籽油的加氢脱氧,结果发现在420℃时,催化剂可以实现菜籽油的100%转化,且HDO反应路线的选择性接近95%。作者认为高转化率与Sn氧化物上的氧空位对羧酸基团的吸附和活化相关,而高HDO选择性则可能是由于催化剂表面的PtSn3±δ金属互化物对H2分子的吸附和活化所致。Murata等[36]研究了不同Re负载量的PtRe/HZSM-5催化剂上的麻疯树籽油的加氢脱氧,结果发现Re的加入大大提高了催化剂的催化脱氧活性。在高质量空速(10 h-1)下,双金属PtRe/HZSM-5(Re/Al物质的量比值0.8)催化剂上C15~C18烷烃产率可达到67%,而单金属Pt/HZSM-5 催化剂上C15~C18烷烃产率仅为2.3%。此外,PtRe/HZSM-5催化剂上的反应路线以HDO为主。
2.4 一步法实现加氢脱氧和临氢异构
动植物油脂经过加氢脱氧生成的产物主要为碳原子数为15~18的直链正构烷烃,这些烷烃产物虽然具有高的十六烷值(约为100),但低温流动性能却很差,因而限制了其应用。进一步将直链烷烃异构化生成支链异构体可以有效改善其低温流动性能[37]。Neste Oil公司工业化了这种两段分开的加氢工艺:动植物油脂先加氢脱氧生成正构烷烃,正构烷烃再临氢异构化生成支链异构体。然而,这种两段加氢工艺势必会增加投资成本和降低工艺效率,如果能够一步实现加氢脱氧和临氢异构双重功能,将具有更大的吸引力。近年来,一步法将动植物油脂加氢脱氧处理制备异构烷烃得到了广泛研究。
直链烷烃异构化通常由具有金属和酸双功能的催化剂催化。金属功能通常为Pt、Pd、Ni等具有较强氢活化能力的金属提供,而酸功能则由Al2O3、SiO2-Al2O3、分子筛等酸性载体提供。为了改善动植物油脂加氢脱氧产物的低温流动性,早期的研究采用了ZSM-5[38]、SiO2-Al2O3[39]、HY[40]等作为催化剂载体,由此得到的催化剂虽然可以获得一定的异构烷烃收率,但是由于酸性太强,长链异构烷烃进一步发生严重裂化副反应,生成中短链的烃类产物(C1~C14),大幅降低了液体产物的收率。随后,研究兴趣逐渐转移到具有中等酸性和一维十元环孔道结构的SAPO-31、SAPO-11、ZSM-22等分子筛载体。这些载体材料除了其中等酸性强度可以抑制裂化反应外,还拥有一维十元环孔道结构,这种孔道结构在长链烷烃异构化反应过程中可以通过其形状选择性在孔道内有效限制多支链异构体的生成,从而进一步抑制裂化反应的发生(多支链异构体更容易发生裂化反应)。Kikhtyanin等[41]研究了Pd/SAPO-31催化剂上葵花籽油的加氢转化,结果发现在较高反应温度和较低空速条件下可以获得较高的异/正烷烃比值(2~16),但该条件下裂化反应也很严重(9.3%~72.1%)。催化剂的高裂化活性可能归结于其加氢/酸功能不平衡。如果载体的酸功能远强于金属的加氢功能,碳正离子中间体将发生裂解而不是骨架异构化。Chen等[15]研究了Pt/SAPO-11催化剂上麻疯树籽油的加氢转化,结果发现Pt/SAPO-11催化剂具有极高的加氢脱氧和异构化活性,其中Si/Al物质的量比值为0.4,Pt含量为3%的样品性能最佳,iso-C15~18的收率高达83%。Pt/SAPO-11催化剂上的加氢脱氧路线以DCO路线为主,C17/C18比值为1.1~1.2。Chen等[42]研究了PtSn/SAPO-11催化剂上棕榈酸甲酯的加氢脱氧,结果发现PtSn/SAPO-11催化剂的活性优于Pt/SAPO-11催化剂。与单金属Pt催化剂不同,双金属PtSn催化剂上加氢脱氧路线以HDO路线为主,产物主要为C16烃类。Sn/Pt的原子比值为2时,催化剂具有最佳的加氢脱氧和异构化性能。作者将高加氢脱氧和异构化活性归结于Pt-Sn合金的形成,而加氢脱氧途径的改变则归结于催化剂上SnO2-x的出现。Smirnova等[43-44]研究了一系列Pt/SAPO-31催化剂上葵花籽油的加氢处理,结果发现Pt/SAPO-31催化剂具有极高的异构化活性,异构烷烃收率高达75%。长期试验中发现450℃煅烧的催化剂样品在连续运行45 h后未发现失活现象,具有较高的活性稳定性。这可能是由于Pt具有较高的加氢功能,与载体SAPO-31的酸性达到一个较好的平衡。Chen等[45]研究了一系列核壳结构的Pt/ZSM-22@SiO2催化剂上棕榈酸甲酯的加氢脱氧,结果发现SiO2壳层可以有效覆盖ZSM-22分子筛外部酸性位点,从而显著提高单支链异构产物的收率,获得高十六烷值的液体产物。但是随着SiO2壳层厚度的增加,催化剂的脱氧活性和异构化选择性逐渐降低。这可能与SiO2包覆一方面会致使Pt颗粒暴露出Pt(100)面比Pt(111)面更多,另一方面会使催化剂的Brφnsted酸中心数量减少有关。而降低催化剂核层ZSM-22的SiO2/Al2O3比可以提高催化剂的催化性能。SiO2/Al2O3比越低,催化剂上的Pt颗粒越小,暴露出的Pt(111)角位越多。
从上述研究可以发现,通过加氢脱氧和临氢异构化反应可以得到高异/正构比例的液体产物,然而这些研究中并没有给出液体产物的低温流动性能。图5比较了碳原子数为12~20的相同碳原子数的正构烷烃、2-甲基异构烷烃和5-甲基异构烷烃的凝固点。由图5可以发现,即使是将直链烷烃异构成为单支链的5-甲基异构烷烃,凝固点也可以下降44℃以上,使液体产物的低温流动性得到大幅改善。

注:ΔT为正构烷烃异构化生成5-甲基异构烷烃的凝固点差图5 不同碳原子数正构烷烃及其异构体的凝固点
2.5 催化剂失活
贵金属催化剂失活的主要原因通常有中毒、结焦和金属烧结[46]。由于甘油三酯类化合物通常含有不饱和键,且加氢脱氧的温度相对较高,反应过程中可能易发生结焦现象。Zhou等[27]在研究微藻油加氢脱氧过程中发现,硫化NiMo/Al2O3催化剂上焦炭的生成量随时间的延长而逐渐增加,然而,Pt/Al2O3或Rh/Al2O3催化剂上只发现极少量的结焦。这可能主要是因为贵金属催化剂具有高氢解离能力,可以通过使焦炭前体物发生加氢反应来抑制焦炭的生成。与结焦相比,金属烧结是贵金属催化剂更常见的失活原因。研究表明,当还原温度高于400℃时,贵金属会发生烧结[23]。另外,甘油三酯类化合物加氢脱氧过程中生成大量的水则会显著提高烧结速率。Jeong等[47]研究了Pt/Al2O3催化剂上棕榈油的加氢脱氧,结果发现Pt/Al2O3催化剂虽然具有较好的催化活性,但在运行36 h后催化剂活性明显降低。作者认为是因为催化剂上的Pt颗粒的烧结和炭沉积引起Pt活性中心减少所致。He等[28]在研究Ru/TiO2催化剂催化脂肪酸酯的加氢脱氧时发现了Ru的流失,导致催化剂出现小幅的失活。其他研究也报道了贵金属催化剂的这种失活[23, 41]。为了防止贵金属烧结和流失,可以通过添加其他金属物种形成合金或采用合适的金属氧化物载体来稳定贵金属。但是,贵金属催化剂也容易受硫、氮和磷化物影响而中毒。据报道毫克每千克水平的硫和氮就可以使单贵金属催化剂中毒,但也可以通过添加第二金属组分来提高催化剂对这些毒物的耐受性[10, 48]。
3 结 语
迄今为止,关于贵金属催化剂上动植物油脂加氢脱氧的研究,科研工作者们做了大量的工作并取得了一定的成果:Pt/Nb2O5、Pd/Nb2O5-SiO2、Ru/TiO2和Ru/C等催化剂显示了极高的低温脱氧活性,而PtSn/SAPO-11、Pt/SAPO-31等催化剂则显示了较好的脱氧和异构双重功能。然而,在催化剂的设计、制备方法、反应机制等方面研究尚存在许多不足,主要有:①在催化剂结构对反应机制、脱氧路径和催化剂失活的影响等方面研究不够深入;②对活性金属与载体之间的协同效应和相互作用,以及双活性金属之间的合金效应等方面研究不够充分;③在如何降低贵金属含量,提高催化剂活性和热稳定性等方面研究尚存在不足。
为解决上述问题,未来的研究工作可以考虑从以下两个方面开展:①在催化剂开发方面,由于贵金属的成本较高,大规模工业化应用的前提是必须大幅降低催化剂的贵金属含量,同时催化剂的活性和稳定性维持在较高水平。为实现这一目标,一方面可以通过引入第二活性金属与贵金属之间形成合金效应,提高催化剂的活性和稳定性;另一方面可以采用合适的载体及制备方法,在载体与活性金属之间产生强相互作用,提高活性金属的分散度和稳定性。②在反应机制和构效关系研究方面,采用科学的催化剂设计与先进的原位表征手段相结合,深入了解反应过程催化剂结构和反应中间产物的变化并解释反应机制,进一步为新型高效催化剂的设计和反应机制的研究提供理论指导。
