全蝽线粒体全基因组序列及其进化分析
2022-09-19高晓云魏久锋丁晓飞刘迎香赵清
高晓云,魏久锋,丁晓飞,刘迎香,赵清
(山西农业大学 植物保护学院,山西 太谷 030801)
昆虫线粒体基因组由一个长度为15~20 kb的闭合环状双链DNA分子组成[1],通常编码37个基因(13个蛋白质编码基因(PCGs)、22个转运RNA基因(tRNAs)、2个核糖体RNA基因(rRNAs))和1个控制区域[2]。与核基因相比,线粒体基因组小且多态,进化速度快,很少进行基因重组[3]。因此,许多研究者用它鉴定物种并进行系统发育关系重建[4-6]。在过去的10 a中,随着测序技术的高速发展,线粒体基因组测序量显著增加,被广泛应用于系统地理学、分子进化和系统发育领域[7-8]。
蝽总科是异翅亚目中最大的总科之一,广泛分布于世界各地,大约包含8 000多个物种[9]。近年来,许多学者研究了蝽总科的系统发育关系,试图为蝽总科的分类提供更多的见解。例如,Gapud首次通过系统发育方法研究了蝽总科,并利用形态学数据推断了各科间的系统发育关系[9]。在不同的分析方法下,蝽总科被认为是单系的[7,10-12]。GRAZIA等学者根据分子数据集[11-14]或形态学[9,14]认为,异蝽科是蝽总科中其他科的姐妹群[14]。在随后的研究中,其他人使用部分数据来推断兜蝽科[15]和荔蝽科之间的关系[15-16]。后来,越来越多学者经常发现兜蝽科和荔蝽科之间的姐妹关系[11-13,17-19]。研究发现,蝽总科内除了兜蝽科和荔蝽科之外,其他科级关系混乱复杂,尤其是蝽科的位置[19-20]。比如,1991年Gapud基于形态构建的蝽总科系统发育树中蝽科与土蝽科是姐妹关系;2008年GRZAIA等[14]通过形态特征和COI、12S、16S、18S、28S相结合证实了部分Phloeidae和蝽科是姐妹群,随后仅基于形态学却发现Lestoniidae和蝽科的姐妹关系;2010年KMENT等[16]指出,蝽科和龟蝽科是姐妹群;2017年LIS等[17]基于18S和28S,证实了蝽科和(兜蝽科+荔蝽科)是姐妹关系;2019年有学者指出,蝽科和盾蝽科是姐妹组[7];2021年XU等[18]基于线粒体基因组构建的系统发育关系发现,蝽科和(异蝽科+同蝽科)是姐妹关系。
本研究首次对全蝽的完整线粒体基因组进行了测序、组装及注释分析,同时对线粒体基因组和其他蝽总科物种进行了核酸组成、密码子使用、基因顺序以及tRNA二级结构的比较分析,研究结果可进一步丰富蝽科线粒体基因组学数据,通过全蝽线粒体基因组序列并联合其他蝽总科物种的线粒体基因组数据,确定全蝽在蝽科系统发育关系中的位置,并讨论分析蝽科在蝽总科中的分类地位。
1 材料和方法
1.1 标本采集与测序
本试验所用的全蝽标本于2019年8月采自安徽省安庆市岳西县坪鹞落村,并在提取DNA之前储存在100%乙醇中;证据标本(编号SXAU2-6)保存在山西农业大学昆虫学研究所。
取全蝽的胸部和腿部肌肉组织,将其研磨成粉末状后严格按照基因组DNA提取试剂盒中使用手册(德国Hilden Qiagen)进行基因组的提取,使用分光光度计检验合格后,送至百迈克生物技术公司(中国上海)测序。全蝽的线粒体全基因组上传至GenBank(登录号:OL884612)。
1.2 序列的拼接、注释与分析
利用Geneious R9软件进行序列的组装,并对组装后的完整线粒体基因组序列进行注释。13个蛋白质编码基因由开放阅读框查找工具ORF依照无脊椎动物线粒体遗传密码子进行查找,并通过与其他昆虫同源基因的比对来确定每个蛋白质基因的起始和终止密码子的位置。利用MITOS Web(http://mitos.bioinf.uni-leipzig.de/)预测22个tRNAs的位置和二级结构。2个rRNAs基因的边界通过与其他已知的蝽科rRNA基因的比较来确定。
蛋白编码基因的密码子使用情况和核苷酸组成通 过Mega v6.0进行了 统计[21],AT和GC偏斜 通过公式(1)和(2)进行计算。同时,利用DnaSP v5.0计算每个非同义位点的非同义替换数(Ka)、每个同义位点的同义替换数(Ks)。
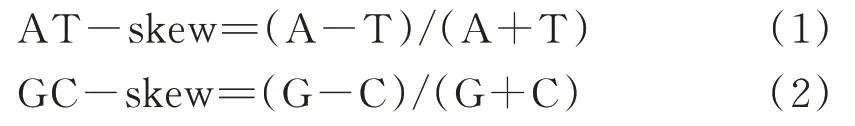
1.3 系统发育树的构建
基于13个蛋白编码基因PartitionFinder得到的分区和模型如表1所示。
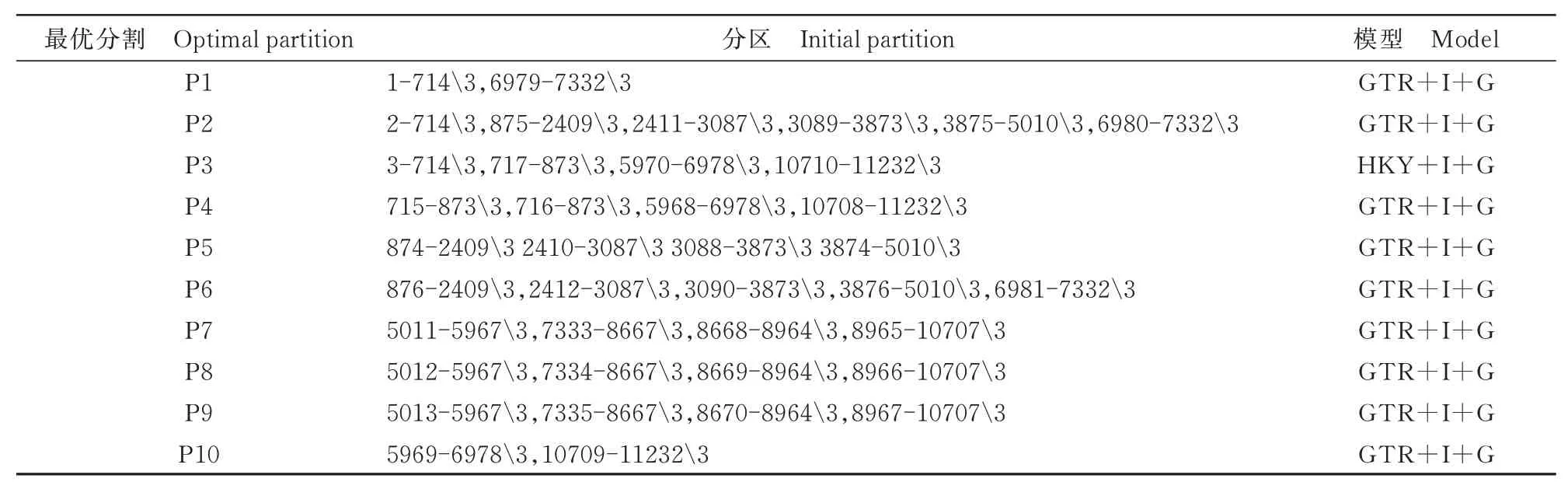
表1 基于13个蛋白编码基因PartitionFinder得到的分区和模型Tab.1 Partitions and models based on Partition Finder of 13 protein-coding genes
从数据库下载9科27种的线粒体全基因组序列,分别基于13个蛋白编码基因和13个基因密码子前2位,进行多重比对,利用PartitionFinder v1.1.1确定划分数据的最佳分区及替换模型。系统发育树法的构建选取最大似然法(ML)和贝叶斯法(BI)。ML树在RAxML-7.0.3中生成,通过dos环境输入命令,命令中需指定外群名称和分区文件,1 000次快速自举检验分析。BI树在MrBayes 3.2.2中运行计算,将PartitionFinder中的分区信息(表1和表2)加到建树命令末尾,以随机起始树开始运算。参数设置为:ngenerations=2 000 000,samplefreq=1 000,printfreq=1 000。当收敛值低于0.01时,程序将停止运行。
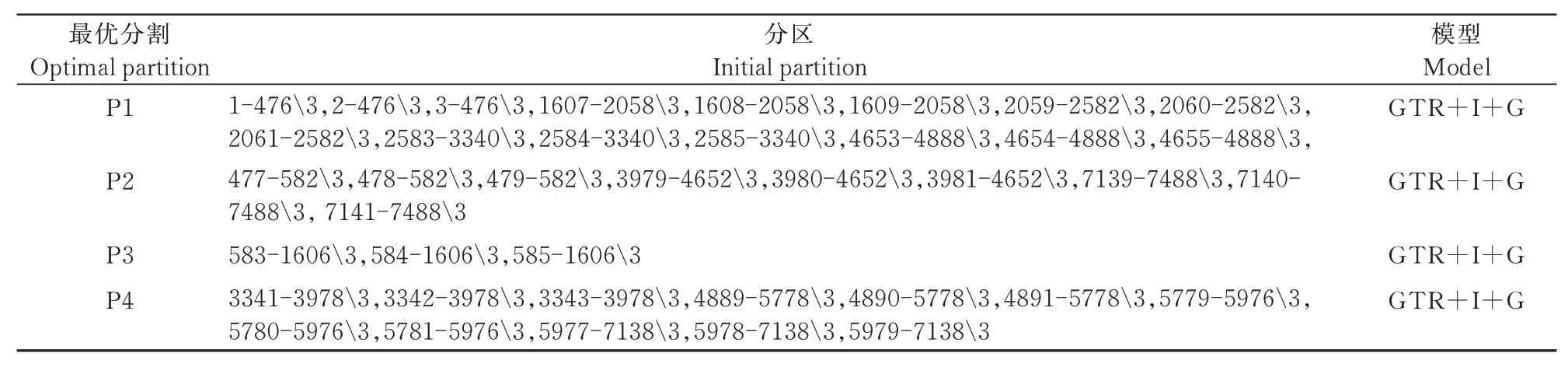
表2 基于13个蛋白编码基因的第一位和第二位密码子PartitionFinder得到的分区和模型Tab.2 Partitions and models based on Partition Finder of first codon and second codon of 13 protein-coding genes
2 结果与分析
2.1 线粒体基因组结构
全蝽的线粒体基因组序列是一个15 479 bp的环状DNA分子(图1)。这个线粒体基因组有37个基 因,包 括2个rRNA、22个tRNA和13个PCG(Protein Coding Gene),其基因顺序与亚库巴果蝇Drosophila yakuba一致(表3)。37个基因中有23个基 因 由J链 编 码(trnI、trnM、nad2、trnW、cox1、trnL2、cox2、trnK、trnD、atp8、atp6、cox3、trnG、nad3、trnA、trnR、trnN、trnS1、trnE、trnT、nad6、cob、trnS2),14个 基 因 由N链 编 码(trnQ、trnC、trnY、trnF、nad5、trnH、nad4、nad4L、trnP、nad1、trnL1、rrnL、trnV、rrnS)。全蝽的线粒体基因组结构紧凑,基因重叠长度范围为1~8 bp,有7处重叠。其中atp8/atp6和nad4L/nad4这2对基因的重叠均为ATGATAA 7个核苷酸。rrnS和trnI之间的非编码区长度为750 bp,在trnA和trnR之间也有一个较大的非编码区。
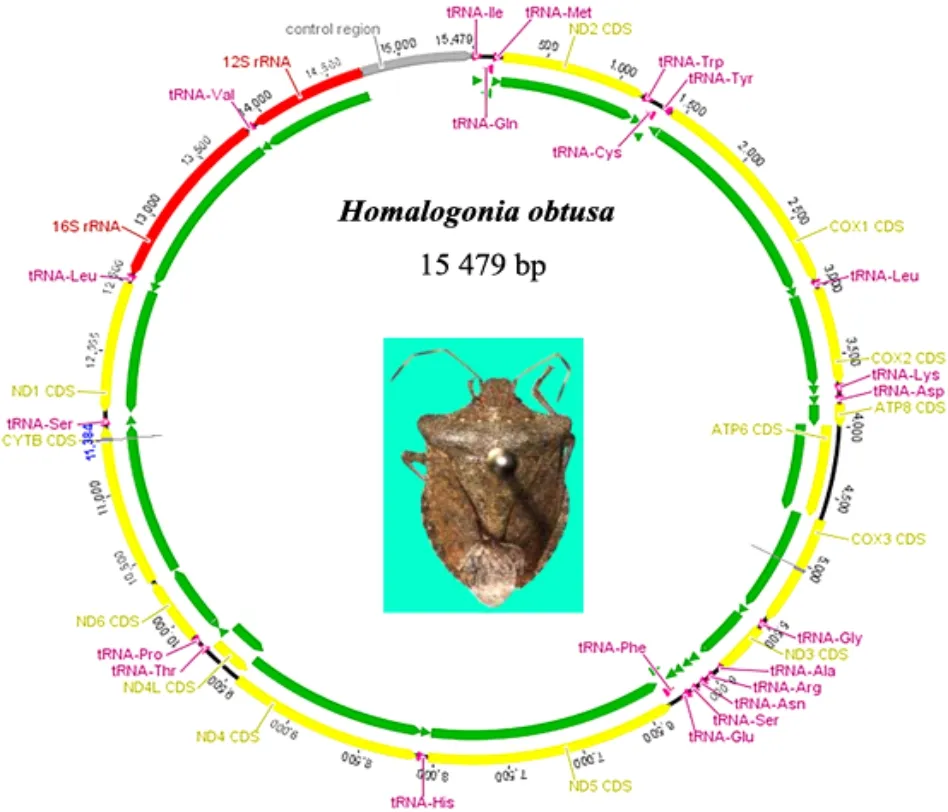
图1 全蝽的线粒体基因组环状结构Fig.1 Circular structure of the mitochondrial gemome of Homalogonia obtusa
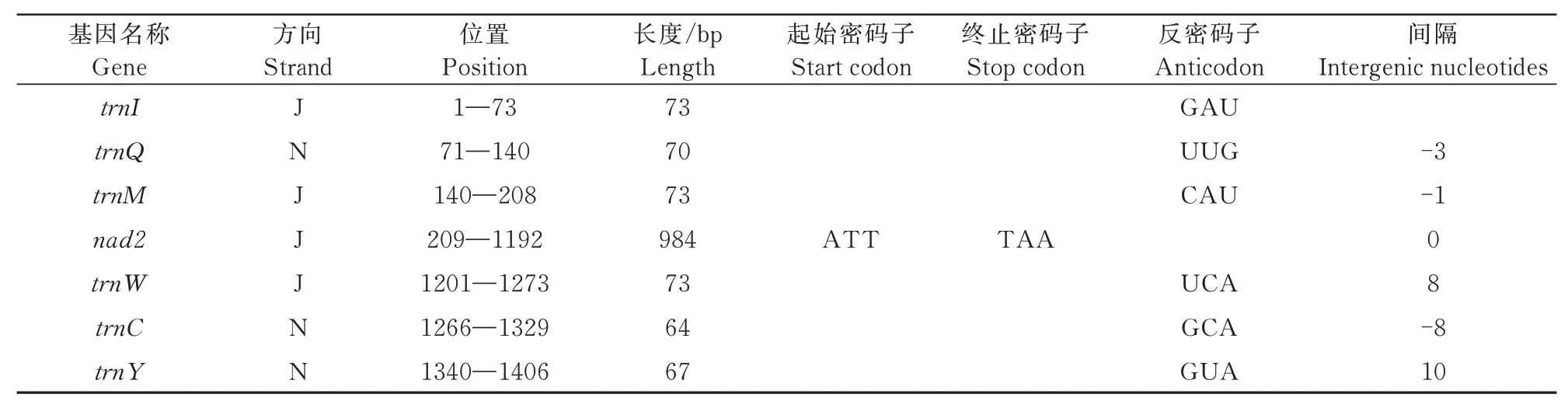
表3 全蝽线粒体基因组结构Tab.3 Structure of mitochondrial genome of Homalogonia obtusa

续表3全蝽线粒体基因组结构Tab.3(Continued)Structure of mitochondrial genome of Homalogonia obtusa
2.2 碱基组成与蛋白编码基因
从表4可以看出,全蝽线粒体基因组的AT含量为76.18%,AT-skew为0.108,具有明显的AT偏好性,位于编码N链蛋白质编码基因中T偏斜值较大,而J链的T偏斜值较小。PCGs、tRNA、rRNA的AT含量均值分别为75.40%、77.70%、78.70%。

表4 全蝽线粒体基因核酸组成情况Tab.4 Nucleic acid composition of mitochondrial gene of Homalogonia obtusa
13个蛋白编码基因中除cox1基因AT含量为69.08%外,其余12个蛋白编码基因中的AT含量均高于70%,且偏向性最大的均出现在密码子第2位。全蝽的13个蛋白编码基因中AT含量最高的3个基因依次是atp8、nad6和nad4L;AT含量比较低的蛋白质基因是cox1、cox2和cox3,故推测高AT含量的基因进化速率快于低AT含量的基因,即atp8的进化速率最高,而cox1的进化速率最低。
昆虫mtDNA的高A+T含量在很大程度上影响了蛋白密码子的使用,本研究中对同义密码子使用情况进行了分析。全蝽中主要是以T或A为第3位的同义密码子的出现频率远高于其他密码子。全蝽的13个PCGs中,A+T含量高于C+G含量,密码子的使用也反映了核酸组成的偏斜。由同义密码子使用度发现,最常用的密码子是NNA,最不常用的密码子是NNG(图2)。在全蝽中最常用的4个氨基酸密码子是UUA(亮氨酸),其次是AUU(异亮氨酸),再次是UUU(苯丙氨酸),最后是AUA(甲硫氨酸)。
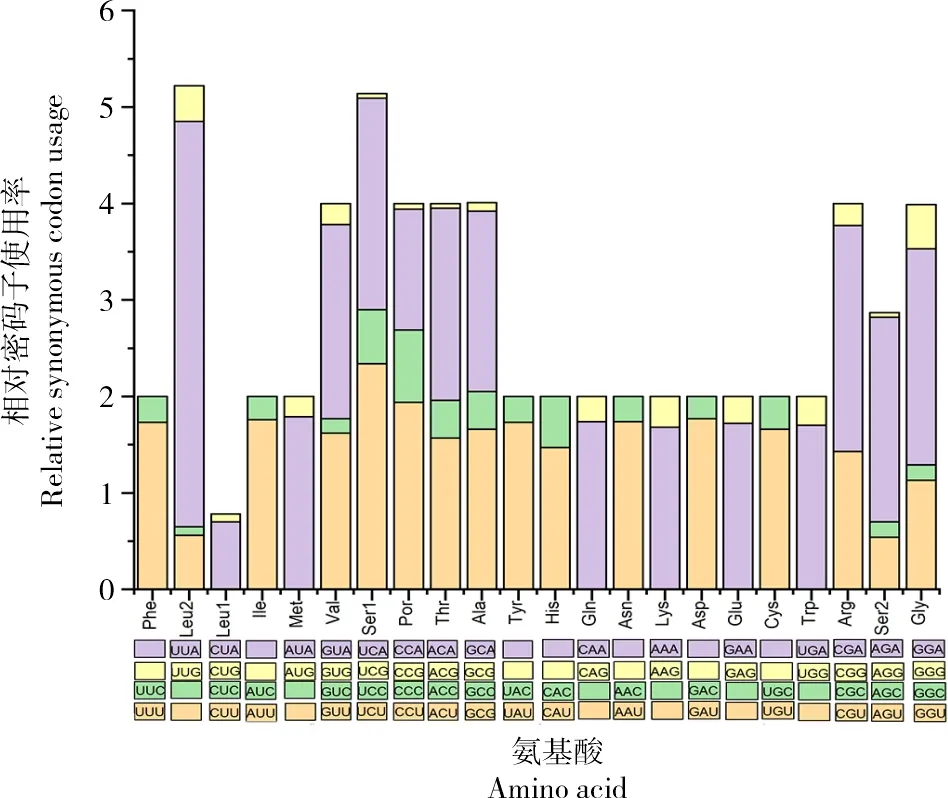
图2 全蝽的相对密码子使用率Fig.2 The relative synonymous codon usage(RSCU)in Homalogonia obtuse
在全蝽的起始密码子和终止密码子中除cox1和nad1以TTG启动外,其他基因起始密码子均为ATA和ATG(ATN)。nad2、atp8、atp6、cox3、nad4、nad5、nad6、nad4l、nad1、cob等10个蛋白质基因的终止密码子是TAA,另外3个cox1、cox2、nad3以单个T终止。起始密码子和终止密码子也主要由A和T组成,这与蝽科其他昆虫的情况一致[11]。
对于单个蛋白质编码基因,本研究计算了它们的同义位点的非同义替代率(Ka)和同义替代率(Ks)(图3)。在核苷酸水平上coxl、cytb、cox3和cox2这4个基因最为保守,进化最慢。非同义与同义置换率的比率(ω=Ka/Ks)在13个基因间变异显 著,coxl、cytb、cox3和cox2偏 低(0.13~0.26),nad1、atp6和nad3中等(0.34~0.35),nad4L、nad5、nad4、nad6、nad2和atp8最高(0.51~0.83)。这与通常认为atp8基因是线粒体基因组中进化最快的观点一致,表明这些基因承受不同的选择性功能约束。然而所有蛋白基因的ω值均小于1,表明这些基因的进化受到纯化选择作用。

图3 蝽总科的13个蛋白质编码的进化速率Fig.3 Evolutionary rates of 13 protein-coding genes in Pentatomoidea
2.3 tRNAs基因 与rRNAs基因
全蝽线粒体基因组中共有22个tRNAs基因,可携带20种氨基酸,其中trnL存在2个不同的tRNA受 体TAA和TAG,trnS存 在2个 不 同 的tRNA受 体GCT和TGA。22个tRNAs基因大多数可以折叠成三叶草状的二级结构(图4)。与大多数报道的蝽科昆虫线粒体基因组一样,trnS1(GCU)的DHU臂被一个简单的环取代,特殊的是,在全蝽中trnA没有TΨC环和TΨC臂。同时,在绘制全蝽的22个tRNAs的二级结构时发现,tRNA臂结构在维持tRNA二级结构稳定中起着关键性的作用,因为它们的核苷酸序列更为保守。tRNA臂连接处以及额外环最为多变,二者都为进化速度比较快的区域。

图4 全蝽的22个tRNA的二级结构Fig.4 Secondary structure of 22 tRNA of Homalogonia obtusa
rrnL和rrnS的长度分别为1 278、816 bp;其中,rrnL位于trnV和trnL(TAG)间隙,rrnS位于trnV和控制区间隙。在rrnL的二级结构中有6个结构域,其中第3结构域缺失,rrnS的二级结构中有3个结构域。
2.4 系统发育分析
本研究以2条长蝽科的物种序列作为外群、蝽总科26个的物种作为内群,基于贝叶斯推断BI(图5、6)和最大似然法ML(图7、8),进行蝽总科内的系统发育分析。选择13个PCGs和13个PCGs的第1位和第2位密码子(PCG12)串联序列数据集构建系统发育树。
基于13个PCGs在贝叶斯分析中,蝽总科内的关系是(异蝽科+(蝽科+(同蝽科+((龟蝽科+盾蝽科)+(土蝽科+(兜蝽科+荔蝽科)))))),这与基于PCG12在贝叶斯分析得到的拓扑结构一致;在最大似然法分析中,基于13PCGs的蝽总科内的关系是(异蝽科+(同蝽科+(蝽科+((龟蝽科+盾蝽科)+(土蝽科+(兜蝽科+荔蝽科))))));基于PCG12的蝽总科内的关系却是(蝽科+((异蝽科+同蝽科)+((龟蝽科+盾蝽科)+(土蝽科+(兜蝽科+荔蝽科))))))。
4种拓扑结构的结果均支持全蝽隶属于蝽科,且对于盾蝽科和龟蝽科之间的姐妹群关系、兜蝽科和荔蝽科的姐妹群关系均具有很高的支持率。蝽科的单系性也得到很好的支持。4种拓扑结构的不同之处主要是蝽科与同蝽科和异蝽科之间的系统发育关系。贝叶斯推断结果显示(图5、6),蝽科进化比同蝽科早,但是最大似然法分析得到的结果却是同蝽科是继异蝽科后进化最早的科(图7),基于PCG12的分析中(图8),蝽科比异蝽科进化还早。

图5 基于13个蛋白编码基因利用贝叶斯分析构建的蝽总科系统发育树Fig.5 Phylogenetic trees of Pentatomoidea constructed based on 13 protein-coding genes using Bayesian inference
图7 基于13个蛋白编码基因利用最大似然法构建的蝽总科系统发育树Fig.7 Phylogenetic trees of Pentatomoidea constructed based on 13 protein-coding genes using RaxML inference
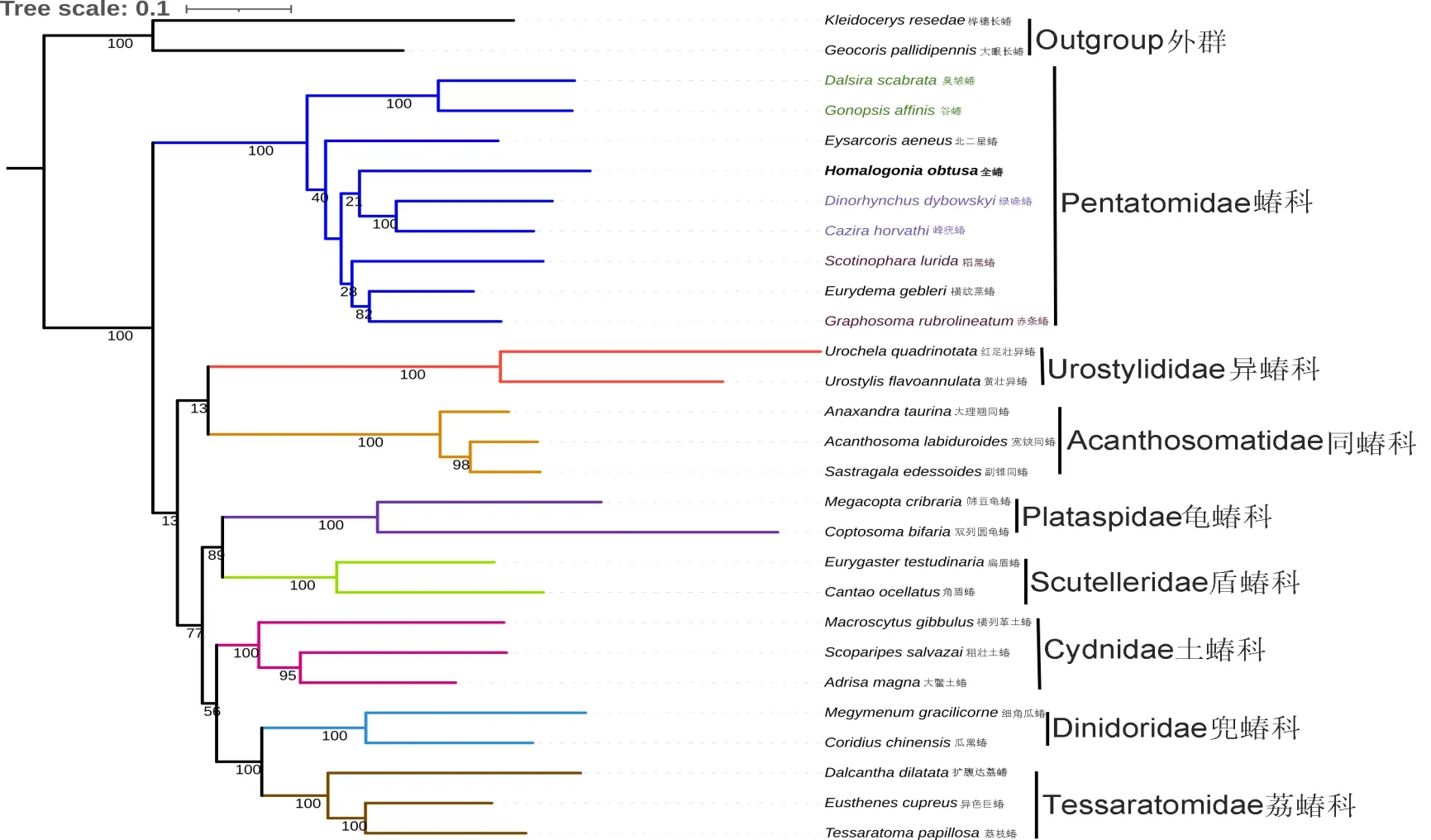
图8 基于13个蛋白编码基因的第1位和第2位密码子利用最大似然法构建的蝽总科系统发育树Fig.8 Phylogenetic trees of Pentatomoidea constructed based on first codon and second codon of 13 protein-coding genes using RaxML inference
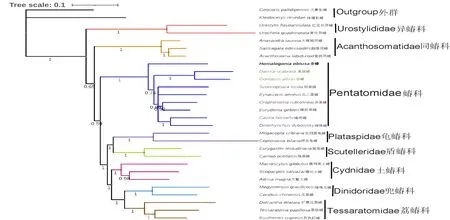
图6 基于13个蛋白编码基因的第1位和第2位密码子利用贝叶斯分析构建的蝽总科系统发育树Fig.6 Phylogenetic trees of Pentatomoidea constructed based on first codon and second codon of 13 protein-coding genes using Bayesian inference
3 结论与讨论
本研究首次对全蝽的线粒体基因组进行了完整测序和报道,线粒体基因组序列的结构和顺序紧凑,与其他蝽科的物种的结构和顺序相同[12,19,22]。通过对全蝽线粒体基因组13个蛋白编码基因的同义密码子使用度、起始密码子和终止密码子及A+T偏斜性等方面分析可以看出,碱基的A+T含量偏斜与进化速率相关。在一些进化较慢的基因中A+T含量比 较 低,比如cox1、cox2和cox3在大多数物种中的A+T含量普遍偏低,其中cox1尤为明显;而在一些进化较快的基因中A+T含量都比较高,如atp8、nad4L和nad6在所选物种中的A+T含量基本偏高。在碱基偏斜方面,N链蛋白基因都是T偏斜度很高,而J链蛋白基因基本为T偏斜但是偏斜度较小。
ATN是13个蛋白编码基因中最常见的起始密码子,此外,TTG也存在于大多数昆虫的线粒体基因组中。在全蝽中,除了cox1和nad1之外,所有蛋白编码基因的起始密码子都是ATN,以TTG开始。全蝽的10个蛋白编码基因的终止密码子是TAA,其余3个蛋白编码基因(cox1、cox2和nad3)的终止密码子是单个T。大多数蛋白编码基因的终止密码子是TAN,然而,一些基因的终止密码子也是单个T或TA[23]。根据研究发现,不完全终止密码子T或TA在转录过程中进行多聚腺苷酸化,最终成为TAA[24]。
本研究中,BI和ML系统发育树的结果均显示,全蝽位于蝽科支系,在分子证据上说明全蝽隶属于蝽科。同时,结果还很好地证明了蝽科的单系性,揭示了蝽科和(龟蝽科+盾蝽科)是姐妹群,这与以前的研究结果一致[18]。文中构建的4棵系统发育树结果显示,蝽科内部的短喙蝽亚科和益蝽亚科是单系性,但是舌盾蝽亚科和蝽亚科是并系,而且蝽亚科内部的物种关系更为混乱。1833年,YANG[25]基于盾片形状将Graphosoma归于蝽亚科Graphosomini,但是其他的研究中都是把它置于舌盾蝽亚科[26]。本研究中,也将Graphosoma归于舌盾蝽亚科,但是它并不与舌盾蝽亚科的Scotinophara聚为一支。造成这样的研究结果可能是因为蝽科物种的形态复杂,变异较大,特征不稳定,对其进行系统的划分比较困难。学者划分蝽科各亚科的标准也仅仅是根据此类昆虫的外部形态的某一个或某几个特征,这并不足以确定系统进化关系;也可能是因为本研究中选取的线粒体基因组基因含量不如核基因多,所以,不能充分表达各亚科之间的进化遗传信息,因而划分的结果会有分歧;当然本研究中也受到了样本不足的限制。所以,要想解决蝽亚科和舌盾蝽亚科的分类关系存在的问题,还需要更多的形态和分子数据构建蝽科内各个亚科的系统发育位置。
本研究通过对全蝽线粒体的全基因组测序和生物信息学分析,发现全椿线粒体基因组全长15 479 bp,AT含量为76.18%。其中,13个编码基因的起始密码子多为ATN和TTG,终止密码子为TAN或单个T碱基,atp8进化最快,cox1进 化 最慢;22个tRNA中trnA没有TΨC环和TΨC臂,trnS1(GCU)没有DHU臂,其他20个tRNA为典型的三叶草结构。系统发育树结果显示,全蝽位于蝽科支系且蝽科为单系。本研究结果丰富了蝽科线粒体基因组学和NCBI数据库的基本数据,为进一步深化蝽科和蝽总科的系统发育关系讨论提供了参考。
