基于GBS简化基因组测序评估3个麦洼牦牛保种群的遗传结构研究
2022-09-16马士龙李小伟李响谢书琼刘益丽唐娇江明锋
马士龙,李小伟,李响,谢书琼,刘益丽,唐娇,江明锋*
(1.西南民族大学畜牧兽医学院,四川 成都 610041;2.青藏高原动物遗传育种资源保护与利用国家教育部重点实验室,四川 成都 610041;3.西南民族大学青藏高原研究院,四川 成都 610041;4.四川省龙日种畜场,四川 红原 624401)
牦牛是非常宝贵的家畜遗传资源基因库。中国拥有极其丰富的牦牛资源,总量有1600多万头,占世界的90%以上,全国形成了18个地方品种和2个培育品种[1],保护好牦牛品种资源对于牦牛产业可持续发展具有重要意义。麦洼牦牛,属于乳肉兼用型地方优良品种,因中心产区是四川省阿坝藏族羌族自治州的红原县原麦洼部落而得名;据调查,麦洼牦牛是由阿坝牦牛引入康北牦牛、果洛牦牛的血缘,经长期选育形成[2];1987年被《四川省家畜家禽品种志》[3]收录,1988年载入《中国牛品种志》[3]。由于早期阿坝州草场超载过牧、饲养管理不足、牛群近亲繁殖等导致麦洼牦牛群体有退化趋势,国家于1953年建立了四川龙日种畜场,成立麦洼牦牛保种场[4],并经长期的生产实践,在2014年组建了纯黑(全身被毛为黑色)、粉嘴(全身被毛为黑色,嘴唇毛色为粉色)和弗洛(全身被毛出生时黑色,逐渐变灰色)保种群[5];其中,弗洛群的产肉性能高于粉嘴群和全黑群,纯黑群的产奶性能高于粉嘴群和弗洛群。近年来,基于性能特征划分的麦洼牦牛群体的生产性能均得到了提高[6],但3个保种群的遗传多样性现状,是否已经出现遗传分化的问题一直未展开相关研究;因此,对麦洼牦牛进行遗传多样性和遗传进化分析,评价不同保种群的保种效果显得尤为重要。
自20世纪以来,国内学者已经通过微卫星标记[7-8]、血液蛋白基因座标记[9]、线粒体DNA标记[10-12]等方法对麦洼牦牛、九龙牦牛、斯布牦牛等品种进行群体遗传进化分析,然而这些DNA标记仅能覆盖部分且少量的牦牛基因组,所取得的基因组遗传变异数据也是非常有限的。而新一代测序技术实现了单核苷酸多态性(single nucleotide polymorphism,SNP)分子标记的高通量检测,可从整个基因组水平上准确高效地研究牦牛的遗传进化关系[13]。基因分型测序(genotyping by sequencing,GBS)技术是在二代测序基础上发展的简化基因组测序技术,通过酶切加tag的方法进行测序,获取酶切位点附近基因序列信息,进而检测大量且准确性高的变异SNP位点信息[14],被广泛应用于遗传图谱构建、全基因组关联分析和群体遗传分析等领域[15-18]。
本研究利用GBS技术研究了麦洼牦牛3个保种群的遗传多样性、群体结构和亲缘关系,评价保种效果,为今后麦洼牦牛遗传资源的保护与利用提供了可借鉴方法和理论依据,为下一步进行麦洼牦牛的良种选育和生产性能提高奠定基础。
1 材料与方法
1.1 试验动物和方法
试验于2020年6月在四川省阿坝藏族羌族自治州红原县国家级肉牛核心育种场—龙日种畜场进行,该地区处于查真梁子山北部区域,海拔3500~4000 m,地势平坦;只有冷、暖季之分,属大陆性高原寒温带季风气候,牧草丰茂,是全国著名的地方优良品种-麦洼牦牛、藏绵羊、河曲马主产区[4]。从3个麦洼牦牛保种群全黑群(QH)、粉嘴群(FZ)、弗洛群(FL)中随机选取406头麦洼牦牛(公∶母=1∶3.5)(表1),群体内个体间血缘关系未知,采集血样样本。具体步骤为用一次性注射器从牦牛颈外静脉采集5 mL血液于乙二胺四乙酸(ethylene diamine tetraacetic acid,EDTA)抗凝管,放入冰盒中带回,放置-80℃冰箱中保存备用。
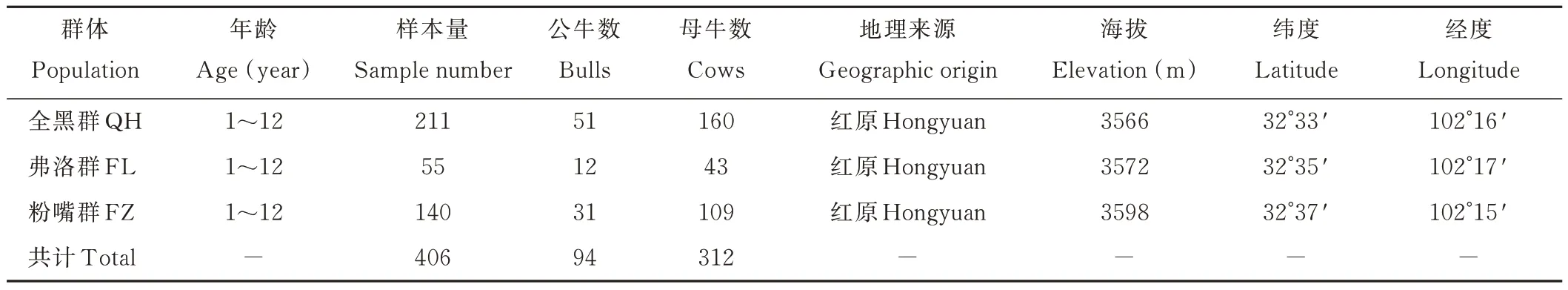
表1 3个保种群的采集信息Table 1 The sample information of 3 breeding populations
1.1.1 主要试剂及仪器 血液基因组DNA提取试剂盒(D3392-01)购自Omega Bio-Tek公司;异丙醇、无水乙醇、琼脂糖、50×TAE、6×Loading Buffer、DNA Marker(BM5000)等购自上海生工生物工程技术服务有限公司和爱必信(上海)生物科技有限公司。
主要仪器有离心机(Eppendorf,5804R,德国)、电子天平(Denver,TP-214,美国)、电泳仪(Bio-rad,美国)、凝胶成像系统(Invitrogen,美国)、紫外分光光度计(Thermo Scientific,NanoDrop 2000,美国);Qubit®2.0荧光计(Invitrogen,美国)。
1.1.2 DNA提取 按照血液基因组DNA提取试剂盒的说明书提取406个血液样本的基因组DNA,利用1%琼脂糖凝胶电泳(100 V,40 min)和紫外分光光度计检测DNA的纯度(A260nm/A280nm值)和完整性,使用Qubit®2.0进行DNA浓度测定,样品纯度要求A260nm/A280nm值为1.8~2.0。
1.2 GBS试验流程
1.2.1 GBS文库构建 文库构建参照董世武等[18]的方法,检测合格的基因组DNA用Mse I限制性核酸内切酶进行酶切,酶切后基因组片段两端加上带有barcode的接头(120 bp)后,利用PCR对每个样品进行扩增,然后对样品进行混合,选择需要的片段(265~315 bp)进行文库构建。
1.2.2 文库库检及上机测序 构建好的文库用Qubit®2.0进行初步定量,具体操作为稀释文库至1 ng·μL-1,再用Agilent 2100检测文库的插入片段大小是否符合265~315 bp,然后为保证文库质量,使用Q-PCR方法对文库的有效浓度进行准确定量(文库有效浓度>2 nmol·L-1)。库检合格后,把不同文库按照有效浓度及每个样本可产生10万个标签的目标下机数据量的需求进行样本混合,即混池,然后进行Illumina Hiseq PE150双末端测序。
1.3 数据统计与分析
1.3.1 测序数据及SNP的质控 Illumina Hiseq测序平台得到的原始图像文件经碱基识别分析转化为测序序列数据(raw reads),这些raw reads需进行质量控制,剔除含有接头序列的reads和低质量reads之后得到的数据即为有效数据(clean data)。
以野牦牛(Bos mutus)(生物项目数据库:PRJNA221623)为参考基因组,通过BWA软件(参数:mem-t 4-k 32-M)[19]将双端有效数据比对到参考基因组,统计平均比对率、平均测序深度和平均覆盖度。采用SAMTOOLS[20]软件进行群体SNP的检测,得到位点变异文件,以测序深度(sequencing depth)为3,缺失(miss)为0.2,次要等位基因频率(minor allele frequency,MAF)大于0.05条件过滤SNP位点,最后获得高质量的SNP位点用于后续分析研究。
1.3.2 群体结构与遗传多样性分析 基于个体间SNP差异,通过GCTA[21]软件计算特征向量和特征值,用R语言绘制第一主成分与第二主成分的主成分分析(principal component analysis,PCA)二维图。使用TreeBest[22]软件中的P-distances模型计算遗传距离矩阵并构建系统进化树,引导值(bootstrap values)设置为1000,检验可靠性。采用ADMIXTURE软件构建群体遗传结构和群体世系信息,假定的祖先群体个数为2~20,根据交叉验证错误值(cross validation error,CV)确定最优K值(一般CV最小的为最优K值)。使用PopLDdecay(v 3.40)[23]进行连锁不平衡分析(linkage disequilibrium,LD),并用R软件绘制LD衰减图。
利用PLINK v 1.90[24]计算观测杂合度(observed heterozygosity,Ho)和期望杂合度(expected heterozygosity,He)等遗传多样性指标。使用R包genepop统计分析每个SNP位点在所有个体中的近交系数(inbreeding coefficients,Fis)和群体遗传分化系数(genetic differentiation index,Fst)。基于筛选后高质量SNP信息,进行选择消除分析,采用PLINK软件进行选择信号检测,以100 Kb为窗口、10 Kb为step进行滑动的核苷酸多样度(π)比率和Fst计算,分别以π比率和Fst值top 10%作为阈值,对群体选择信号进行分析,其top 10%的交集为候选位点,并对受选择位点所在基因进行GO富集分析。
2 结果与分析
2.1 血液基因组DNA提取
提取406头牦牛样本血液基因组DNA,经1%琼脂糖凝胶电泳检测发现,条带清晰明亮(图1),各项参数均符合测序所需标准。

图1 部分样本血液基因组DNA电泳图Fig.1 Genome DNA electrophoresis of some blood samples
2.2 测序数据产出与质控
对406份牦牛血液基因组进行GBS测序和质量控制(详见1.3.1),获得有效碱基数279.211~2456.350 Mb,共产生327.517 Gb原始数据,平均为806.690 Mb;过滤后共产生327.512 Gb有效数据,有效率达99.990%,平均为806.680 Mb。碱基检测错误率为0.040%~0.060%,平均仅有0.044%(表2);达到Q20质量的碱基占比为93.83%~97.58%,平均为95.73%;GC碱基含量为36.56%~40.28%,平均为38.88%;获得有效读长数为1.94~17.08 Mb,平均为5.60 Mb;有效读长比对到参考基因上的占比为92.60%~99.68%,平均比对率为98.75%;测序碱基总量与基因组的比值,即测序深度最小为5.65×,测序深度最大为31.64×,平均测序深度为14.33×,平均覆盖度为3.46%(表3)。表明测序质量高,GC分布正常,样本均没有被污染,此次建库测序成功。

表2 麦洼牦牛测序数据统计Table 2 Summary of sequencing data of Maiwa yak

表3 测序数据质量评估Table 3 Quality assessment of sequencing
2.3 SNP检测
应用SAMTOOLS等软件对测序有效数据进行SNP检测,共鉴定出1628805个SNP位点,进行质控后获得高质量SNP位点126122个。质控后的SNP经过与牦牛参考基因组(生物项目数据库:PRJNA 221623)比对分析,基因组的SNPs数目及基因组序列长度如图2所示,总体上来看SNPs的数目与基因组序列长度总体上呈正相关;序列号为NW_005393843.1的片段最大(8785736 bp),所含SNP数目最多,为463个SNP位点;而由于基因组序列长度差异,不同基因组序列的SNP数目变化较大,NW_005393917.1的 片 段 较 大(542197 bp),却 仅 有1个SNP位点。
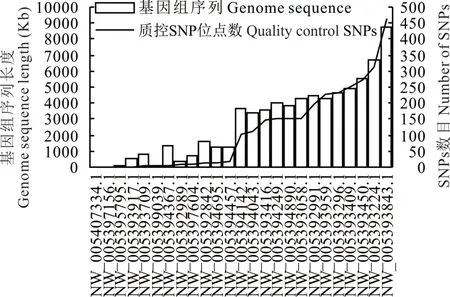
图2 质控SNPs位点数与基因组序列片段长度变化趋势Fig.2 Variation trend of SNPs number and genome sequence length
2.4 遗传多样性和群体结构分析
2.4.1 遗传多样性分析 根据变异位点分析QH、FZ、FL群的杂合度(Ho、He)、核苷酸多样度(π)、近交系数(Fis),结果见表4。3个群的观测杂合度分别为0.3029、0.3042、0.3044,观测杂合度均与期望杂合度接近,只有FZ的观测杂合度稍高于期望杂合度,总体平均观测杂合度与期望杂合度接近。3个群的核苷酸多样度分别为0.3030、0.3020、0.3009,FL群最低;而与QH、FZ群的近交系数相比,FL群是最高的,为0.0209。
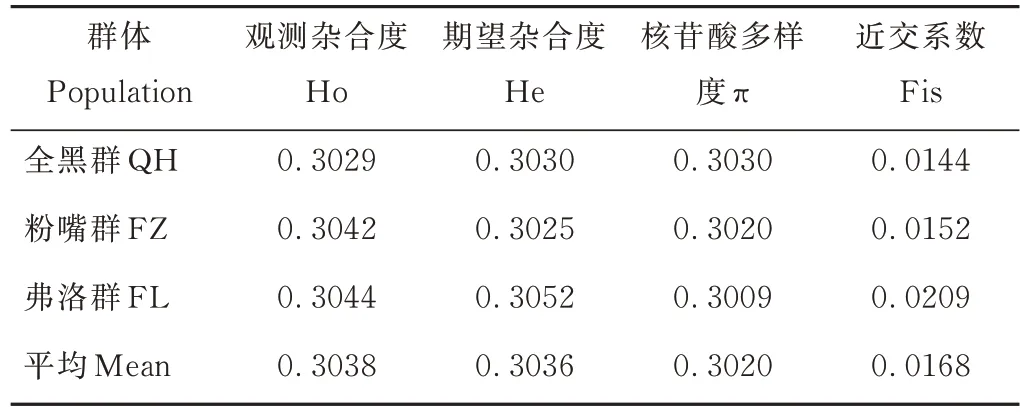
表4 麦洼牦牛3个育种群遗传多样性参数Table 4 Genetic diversity parameters of three preserved populations of Maiwa yak
连锁不平衡(LD)模式见图3,3个群体LD均随距离增长逐渐平滑下降,各等位基因不存在强烈的连锁不平衡水平(r2≤0.5),SNP距离 在0~100 kb的LD衰减 速度较快,LD系数r2约从0.5降至0.2以 下,SNP距离在100~300 kb的LD水平较低。当r2=0.3时,各群体对应LD衰减距离基本相等,说明各群体LD衰减速度差异不大。

图3 LD随距离增长的衰减Fig.3 Attenuation of LD with distance
由表5可以看出,3个群体遗传分化系数Fst为0.02504~0.03513(介于0~0.05),其中QH和FZ群之间的遗传分化程度最大(0.03513),两群的遗传距离同样是最远(0.0358)。
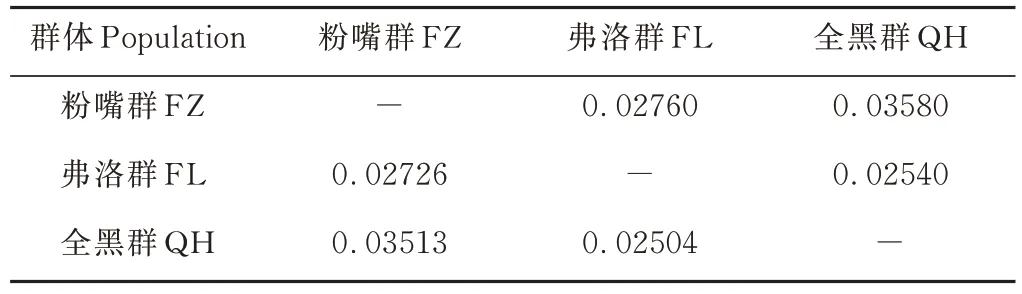
表5 群体间的遗传分化系数(Fst)(对角线下)和遗传距离(DR)(对角线上)Table 5 Genetic differentiation index(below diagonal)and genetic distance(above diagonal)among three populations of Maiwa yak
2.4.2 群体遗传结构和亲缘关系分析 基于个体基因组SNP差异程度,通过PCA二维图的形式来展示个体的聚类情况,可以大致了解群体的遗传结构。由图4可知,麦洼牦牛群体的PC1、PC2的贡献率分别为4.75%、2.72%,FL群紧密聚在一起,与QH、FZ群部分个体关系近;FZ群个体较分散,QH群中有个体偏离群体与FZ群聚在一起,两群存在明显的分层。
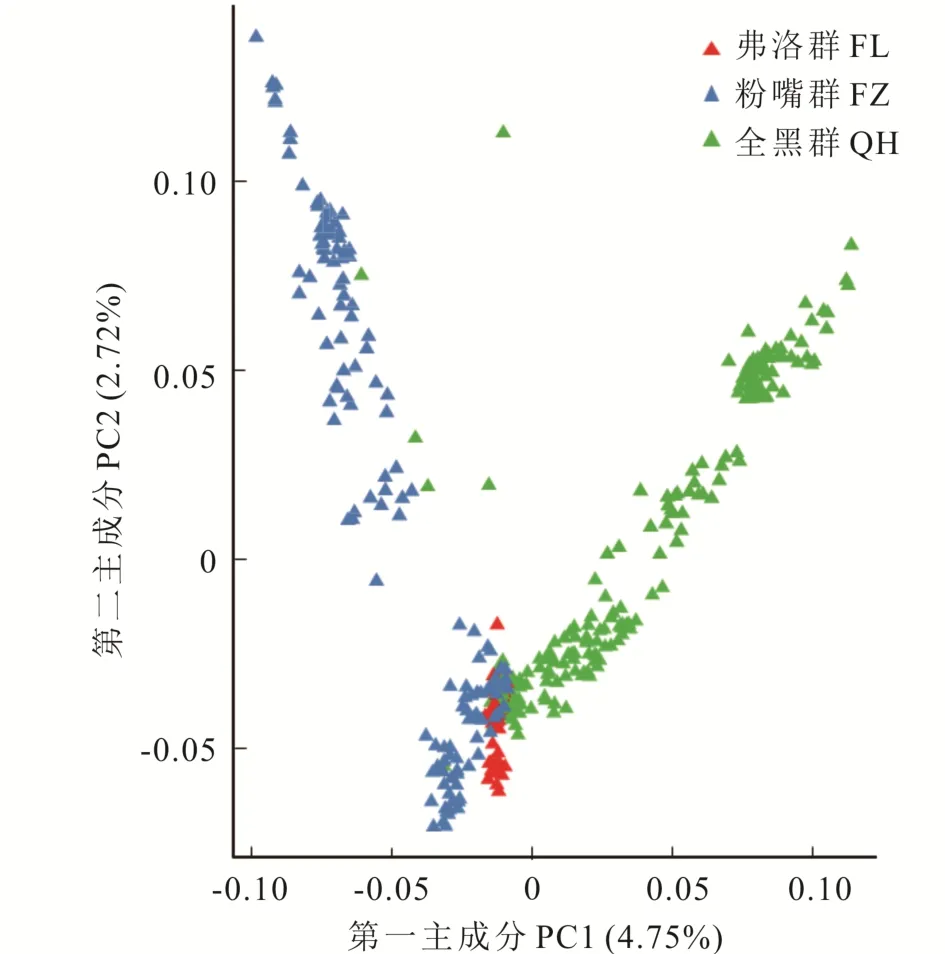
图4 PCA分析二维图Fig.4 PCA analysis of two-dimensional graph
群体遗传结构指遗传变异在物种或群体中的一种非随机分布。按照地理分布或亲缘关系可将一个群体分为若干亚群,处于同一亚群内的不同个体亲缘关系较近,而亚群与亚群之间则亲缘关系稍远。从图5可以看出,3个保种群的血统构成,当假设祖先群为2(K=2)时,QH、FZ、FL群都携带两个祖代血缘;当最 佳K值 为12时 ,FL群 可 分 为 两 个群 ,FZ群 部 分 个体(棕色)可单独划分为一个群,QH群部分个体(粉色)可单独划分一个群,其他需根据系谱记录进行划分。
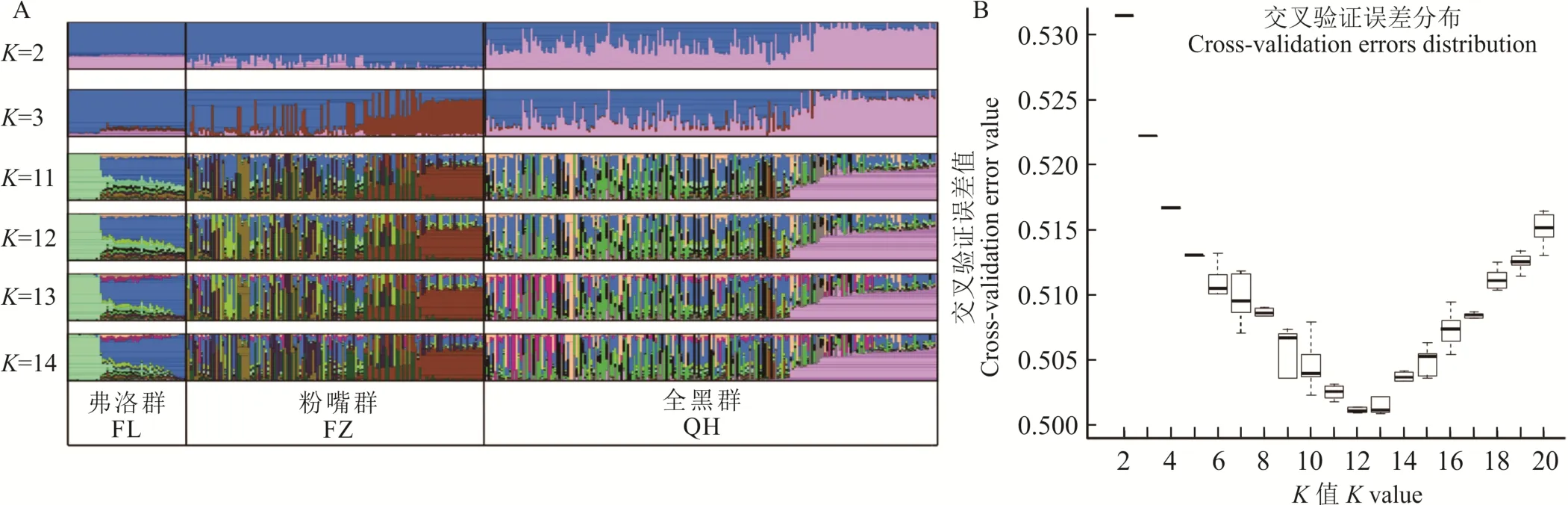
图5 麦洼牦牛structure群体遗传结构Fig.5 Genetic structure of Maiwa yak structure populations
406头麦洼牦牛聚类的系统进化树(图6)直观地以圈图的形式展示它们之间的亲缘关系远近,结果可以看出,QH群聚成一个大支(绿色),群体存在离群个体;FL和FZ群组成另一支,FZ群逐渐分离出来,最后形成一小支(蓝色),FL群有一支最后被分离出来(红色),但整体有一半的个体与QH、FZ群聚在一起。

图6 系统进化树Fig.6 Phylogenetic tree
2.4.3 群体选择信号分析 Fst和π分析是两种有效地检测选择消除区域的方法,特别是在挖掘与生存环境密切相关的基因功能区时可以得到较强的选择信号,便于目标基因的筛选。根据麦洼牦牛群体间Fst和π进行选择信号分析,筛选出3513个差异较大的SNP位点,共有104个差异基因受到选择,进行GO功能注释分析,结果表明(图7),这些受选择基因主要富集在有丝分裂、氯离子运输、节律过程、刺激反应等生物学过程;主要涉及的细胞组成:氯离子通道复合体、细胞间连接、膜的锚定组分等;主要涉及的分子功能包括:氯离子通道活性、酶结合、蛋白质二聚化活性等。KEGG结果富集到了28条通路,主要集中在生殖激素信号、内/外分泌、钙离子信号、cGMP-PKG信号、血管平滑肌收缩等调控通路上(表6)。

表6 受选择基因的KEGG通路Table 6 KEGG pathway of differential expression genes
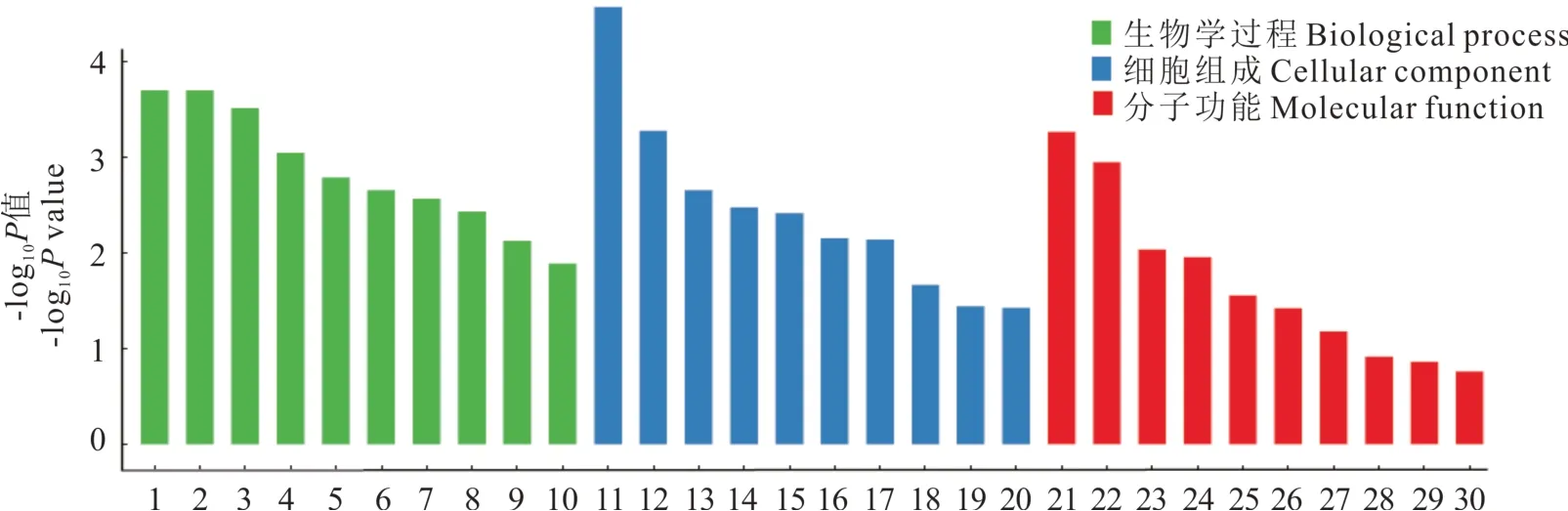
图7 受选择基因的GO富集条目Fig.7 GO enrichment items of selected gene
3 讨论
牦牛是我国青藏高原珍稀而宝贵的遗传资源,在高原畜牧业发展和生态系统平衡方面起着至关重要的作用。牦牛遗传多样性水平和遗传结构的研究不仅是保护牦牛遗传资源的主要内容,也是实施科学有效的品种保护措施的前提条件。随着分子生物技术的快速发展,基于SNP的新一代测序技术成为主流方法,使得从基因组层面上研究牦牛遗传结构和遗传多样性成为可能,更全面精确地监测牦牛保种效果。自麦洼牦牛品种被挖掘以来,其遗传多样性研究是保护这个地方牦牛品种资源的核心问题之一;因此,本研究对406头麦洼牦牛进行GBS测序探究其群体的遗传多样性及遗传结构,测序结果得到了806.68 Mb的有效数据量,共鉴定出126122个SNP位点,这些数据将为未来制备麦洼牦牛育种SNP芯片提供基础。
遗传杂合度是度量群体遗传多样性的最适参数,其观测杂合度和期望杂合度的接近程度可反映群体遗传多样性的高低。本研究中麦洼牦牛3个保种群的观测杂合度为0.3029~0.3044,属于中度杂合群体,整个群的平均观测杂合度与平均期望杂合度接近,这表明麦洼牦牛品种具有较高纯度和丰富的遗传多样性,这与赵芳芳[25]的微卫星法和师方等[11]的随机扩增多态性脱氧核糖核酸(random amplified polymorphic DNA,RAPD)法结果相似;龙日种畜场为高原浅丘地貌,可利用草地面积1.667万hm2,在这样的相对隔离地理环境中麦洼牦牛得到了有效保护,较低的近交水平也说明保种群并没有受外来品种及近交的影响。另外,粉嘴群的观测杂合度高于全黑群,结果与侯孟典等[26]和柴志欣等[27]的报道一致;这表明粉嘴群所经过的长期定向人工选择强度高于全黑群,有研究[5]指出粉嘴个体相较于全黑个体的乳脂率较高,而挤奶量较低,推测粉嘴群已经在乳质等生产性状上受到了强烈的选择。群体间的遗传分化与选育的方式和世代长短有关,四川省龙日种畜场的牦牛原种场采取的开放式群体继代选育法,在世代选育过程中,按需地从麦洼牦牛主产区的选育群引进优良非近亲种公牦牛进行血缘更新[3],使得场内的麦洼牦牛群体内个体间遗传变异度变高;因此粉嘴和全黑群的遗传分化指数和遗传距离较远,分化呈现出不稳定的混乱状态,有部分血缘交叉和个体离群,这提示在保种过程中,应该加强对种公牛的严格选留工作,避免近亲交配的发生。另外,早期的弗洛个体其实并没有从总群中单独划分出来,而是后来依据其毛色特征和产肉性能才组建的[5],这印证了弗洛群是粉嘴群和全黑群的祖代个体杂交所形成的群落的可能;进一步说明弗洛群含有部分全黑群和粉嘴群血统,有助于解释弗洛群的出生毛色变化现象。龙日种畜场内麦洼牦牛群体的遗传结构随着时间的推移也发生了较大变化,推测是由较小的保种群体规模限制导致,群体结构图和系统进化树都表明全黑群大部分个体聚为一类,弗洛群小部分个体聚为一类,这说明全黑、弗洛群的保种效果相对较好,全黑群和弗洛群出现血统较纯正的个体群,这些个体和粉嘴群一样,它们的某些经济性状可能已经得到了固定。
为鉴别3个保种群之间在基因组特定位点上存在的分化,对麦洼牦牛群体104个受到强烈选择的基因进行GO和KEGG分析,发现这些基因广泛参与生殖机能调控、生长发育、刺激反应等生物学相关过程。其中,特异型钙调磷酸酶PPP3CC基因参与精子发生和睾丸成熟,缺失该基因的小鼠精子尾部中段会发生僵直、超活化能力降低[28],该基因也已经在牛体内被检测到[29];钾离子通道宿主因子KCNMA1与牛的繁殖性能相关,可增强动物性行为[30]。神经生长因子NGF能有效促进卵泡形成,通过调控原始卵泡中扁平细胞分化成次级卵泡中包裹卵母细胞的立方形颗粒细胞过程,在牛孤雌胚胎的早期发育起关键作用[31];鸟嘌呤核苷酸结合蛋白GNAQ基因是通过影响小脑分泌激素从而影响动物毛色形成的重要调控基因[32],也参与动物性腺的发育和生殖细胞的成熟[33]。这表明3个保种群在龙日种畜场的人工选择条件下,公母牛的繁殖力均得到了增强,尤其公牛的性行为方面较为明显。肌细胞增强因子MEF2C主要参与调控肌细胞分化,特别是在从胎牛到成年牛发育过程中,介导骨骼肌、心肌和平滑肌的细胞分化[34];小GTP结合蛋白Rho下游效应因子ROCK2对低氧环境诱导的肺动脉高血压发挥着重要作用,并在成肌细胞分化中持续上调表达,参与肌肉形成过程[35];另外,KCNJ3基因与心律发生有关[36],这3个基因可能与麦洼牦牛的高原适应性和肌肉发育调节相关,说明保种群在强烈的人工选择中有向肉质性状方面发生了定向选择的趋势。谷氨酸突触通路中的ITPR2基因是钙离子跨膜转运活性的关键调控因子,内含子区存在动物抗环境压力的潜在靶点[37],这表明保种群在龙日种畜场的天然草场放牧和一定的补饲管理下,群体的抗应激能力也得到了强化。酪氨酸蛋白激酶受体KIT基因是调控哺乳动物白色皮肤及被毛的基因,KIT基因突变会导致动物由花斑到全白不同程度的白色被毛,而牦牛的白色斑点就是由KIT基因在6号染色体上的易位形成的[38],提示该基因可作为弗洛群的灰色表型发生的候选基因;另外,仍有许多受选择功能基因有待进一步研究注释。本研究在基因组水平上进一步说明龙日种畜场内麦洼牦牛不同保种群经历了定向选择,筛选的受选择功能基因对了解麦洼牦牛的适应性变化并获得其优良的经济性状具有非常深远的意义。
4 结论
本研究利用GBS技术揭示了麦洼牦牛3个保种群的遗传多样性和遗传结构,粉嘴群和全黑群有遗传分化趋势,分群的效果得到了体现,3个保种群的保种效果较好,可在世代之间进行比较,进一步分析保种效果。测序出3513个显著SNP位点和104个差异基因;3个保种群在毛色、肉质、繁殖性状上经历了人工选择,这可为麦洼牦牛的保种选育工作提供建议。
