基于HPLC特征图谱结合化学计量学的菊花特征标志物的研究
2022-09-13常相伟赵宏苏张玖捌金传山吴德玲陆兔林
张 伟,吴 瑞,常相伟,赵宏苏,张玖捌,金传山,3,吴德玲,3*,陆兔林*
1安徽中医药大学药学院,合肥 230012;2南京中医药大学药学院,南京 210023;3中药饮片制造新技术安徽省重点实验室,合肥 230012
菊花为菊科植物菊ChrysanthemummorifoliumRamat.的干燥头状花序,主要含有黄酮类、挥发油、有机酸和微量元素等活性部位[1],具有散风清热、平肝明目及清热解毒的功效。《中国药典》2020版按产地和加工方法的不同,收载的菊花品种有“亳菊”、“滁菊”、“贡菊”、“杭菊”和“怀菊”[2]。作为一种药食同源的中药材,菊花具有品种繁多、成分组成复杂的特点,而不同品种的菊花因其生长环境、栽培加工方法等不同,化学成分含量存在很大的差异,且质量参差不齐,导致其临床疗效也有所不同[3-5]。目前,《中国药典》2020版以绿原酸、木犀草素-7-O-β-D-葡萄糖苷及3,5-O-二咖啡酰基奎宁酸3种化学成分含量为评价指标,难以全面地评价菊花药材的质量[6]。
中药特征图谱可以表征中药内在质量的整体变化,越来越多的中药通过建立特征指纹图谱进行质量控制[7-11],其中HPLC特征指纹图谱技术也常用于菊花的品种鉴别和质量控制研究中[12-17]。中药质量标志物(Q-Marker)由专属的、与临床疗效相关的、可被监测的化学成分组成,与中药功能属性等相关,能反映中药安全性和有效性,可被用于构建现代中药整体、系统、量化的质量评价体系[18]。在Q-Marker的基础上,通过整合指纹图谱和化学计量学方法,发现体现药材或者复方中共性特征的成分,建立符合中药特点的多维质量评价体系,可能对中药的科学监管将提供更好的解决方案。
本研究在菊花前期研究的基础上,通过建立不同品种菊花的HPLC特征图谱,结合化学计量学方法筛选出菊花的共有的物质基础作为菊花的特征标志物,同时筛选出不同品种菊花之间显著性差异的特征标志物[19],从而为菊花的整体质量评价、品种鉴别及单品种特征性评价提供科学依据。
1 实验材料
1.1 仪器
Agilent XDB-C18色谱柱(4.6 mm×250 mm,5 μm);岛津LC-2030C 3D高效液相色谱仪;SPD-20AV检测器(日本岛津公司);KQ-400KDE超声波清洗器(昆山市超声仪器有限公司);800C玖蓝多功能粉碎机(永康市玖蓝五金制品有限公司);SHZ-D(Ⅲ)循环水式真空泵(巩义市予华仪器有限责任公司);电子分析天平(AB135-S,METTLER TOLEDO);BS-300+电子天平(上海友声衡器有限公司)。
1.2 试药
绿原酸(批号:MUST-21030304)、隐绿原酸(批号:MUST-21042003)、新绿原酸(批号:MUST-21030108)、3,5-O-二咖啡酰基奎宁酸(批号:MUST-21011110)、异绿原酸B(批号:MUST-21030602)、异绿原酸C(批号:MUST-21030603)购于成都曼思特生物科技有限公司;咖啡酸(批号:DST191030-013,成都德思特生物技术有限公司);芹菜素(批号:PS010177)、芹菜素-7-O-β-D-葡萄糖苷(批号:PS011186)、木犀草素(批号:PS010346)、木犀草素-7-O-β-D-葡萄糖苷(批号:PS000708)、金合欢素(批号:PS011043)、香叶木素(批号:PS010395)、香叶木素-7-O-β-D-葡萄糖苷(批号:PS011578)均购于成都普思生物科技股份有限公司,纯度均大于98.0%。甲醇、乙腈(瑞典OCEANPAK欧普森公司,色谱纯);冰醋酸(上海阿拉丁生化科技股份有限公司,分析纯);超纯水。
1.3 样品
50批菊花样品分别采收于安徽亳州、安徽滁州、安徽黄山、浙江桐乡、河南焦作,经安徽中医药大学俞年军教授鉴定为菊科植物亳菊Chrysanthemummorifolium(Ramat).Tzvel.‘Boju’ cv.nov.(BJ)、滁菊Chrysanthemummorifolium(Ramat).Tzvel.‘Chuju’ cv.nov.(CJ)、贡菊Chrysanthemummorifolium(Ramat).Tzvel.‘Gongju’ cv.nov.(GJ)、杭菊Chrysanthemummorifolium(Ramat).Tzvel.‘Hangju’ cv.nov.(HAJ)、怀菊Chrysanthemummorifolium(Ramat).Tzvel.‘Huaiju’ cv.nov.(HUJ)的干燥头状花序。样品粉碎,过一号筛,备用。来源信息见表1。
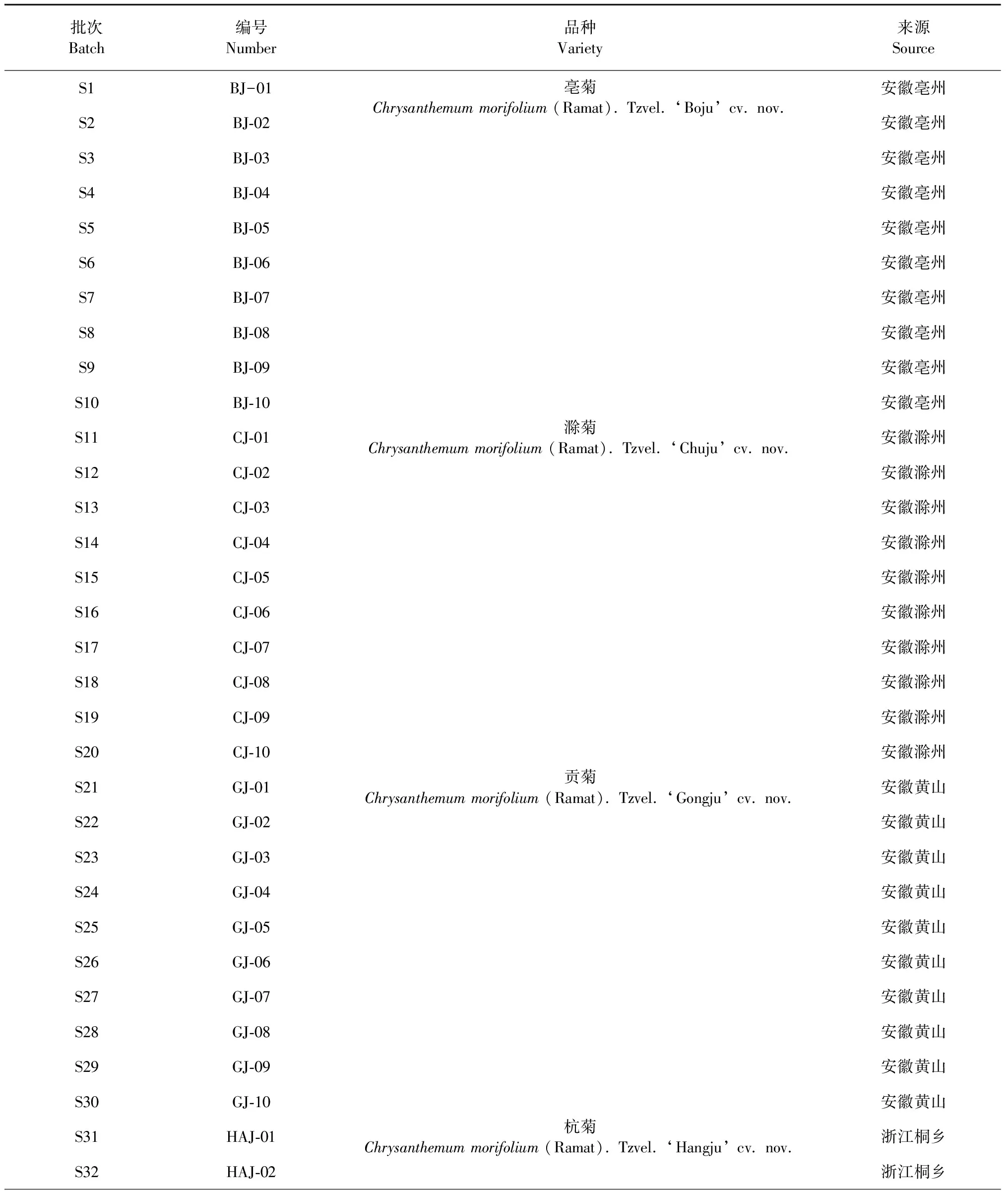
表1 50批菊花样品来源
2 方法与结果
2.1 色谱条件
色谱柱:Agilent XDB-C18色谱柱(4.6 mm×250 mm,5 μm);流动相:乙腈(A)-0.2%醋酸水溶液(B),梯度洗脱(0~8 min,5%→10%A;8~15 min,10%→14%A;15~18 min,14%→17%A;18~22 min,17%→17.5%A;22~35 min,17.5%→18%A;35~40 min,18%A;40~50 min,18%→19%A;50~65 min,19%→30%A;65~80 min,30%→52%A);流速:1.0 mL/min;检测波长:348 nm;柱温:30 ℃;进样量:10 μL。
2.2 供试品溶液的制备
取菊花样品粉末(过一号筛)约0.25 g,精密称定,置具塞锥形瓶中,精密加入70%甲醇25 mL,密塞,称定重量,超声处理(功率300 W,频率45 Hz)40 min,放冷,再称定重量,用70%甲醇补足失重,摇匀,0.22 μm微孔滤膜滤过,取续滤液,即得。
2.3 混合对照品溶液的制备
取绿原酸、木犀草素-7-O-β-D-葡萄糖苷、3,5-O-二咖啡酰基奎宁酸、新绿原酸、隐绿原酸、咖啡酸、异绿原酸B、异绿原酸C、芹菜素-7-O-β-D-葡萄糖苷、香叶木素-7-O-β-D-葡萄糖苷、香叶木素、芹菜素、木犀草素与金合欢素对照品适量,置棕色量瓶中,加70%甲醇制成每1 mL含绿原酸31.2 μg、木犀草素-7-O-β-D-葡萄糖苷23.6 μg、3,5-O-二咖啡酰基奎宁酸86.3 μg、新绿原酸20.9 μg、隐绿原酸22.1 μg、咖啡酸41.5 μg、异绿原酸B 22.7 μg、异绿原酸C 37.8 μg、芹菜素-7-O-β-D-葡萄糖苷59.2 μg、香叶木素-7-O-β-D-葡萄糖苷46.7 μg、香叶木素69.3 μg、芹菜素58.4 μg、木犀草素52.1 μg与金合欢素54.7 μg的混合溶液,即得。混合对照品色谱图见图1。
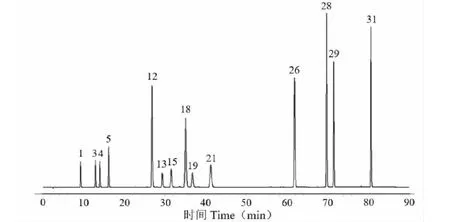
图1 混合对照品色谱图
2.4 方法学考察
2.4.1 精密度试验
精密称取怀菊样品(S41)0.25 g,按“2.2”项下方法制备供试品溶液,“2.1”项下色谱条件测定,连续进样6次,记录特征图谱,计算各共有峰的相对保留时间和相对峰面积。结果各共有峰相对保留时间RSD值(n= 6)为0.04%~0.18%,相对峰面积RSD值(n= 6)为0.01%~1.29%,表明仪器的精密度良好。
2.4.2 重复性试验
精密称取同一批次怀菊样品(S41)0.25 g,6份,按“2.2”项下方法制备供试品溶液,“2.1”项下色谱条件测定,记录特征图谱,计算各共有峰的相对保留时间和相对峰面积。结果各共有峰相对保留时间RSD值(n= 6)为0.06%~0.25%,各共有峰(除18号峰的RSD为5.36%)相对峰面积RSD值(n= 6)为1.24%~4.52%,表明该方法的重复性良好。
2.4.3 稳定性试验
取同一供试品溶液,分别于制备完成后0、2、4、8、12、24 h以“2.1”项下色谱条件测定,记录特征图谱,计算各共有峰的相对保留时间和相对峰面积。结果各共有峰相对保留时间RSD值(n= 6)为0.02%~0.25%,相对峰面积RSD值(n= 6)为0.10%~1.19%,表明供试品溶液在24 h内稳定性良好。
2.5 不同品种菊花特征图谱的建立及分析
2.5.1 五种菊花特征图谱的建立
分别精密称取50批不同品种菊花样品0.25 g,按“2.2”项下方法制备供试品溶液,“2.1”项下色谱条件测定,记录色谱图。将10批亳菊药材的HPLC色谱图以AIA文件格式依次导入“中药色谱指纹图谱相似度评价系统(2012.130723版本)”软件,以S3为参照图谱,多点校正法进行匹配,以中位数法生成亳菊对照图谱(R)。滁菊、贡菊、杭菊、怀菊的对照图谱建立同亳菊。将50批菊花药材的HPLC色谱图依次导入“中药色谱指纹图谱相似度评价系统(2012.130723版本)”软件,生成菊花对照图谱(R)。样品叠加图谱见图2,对照图谱见图3。
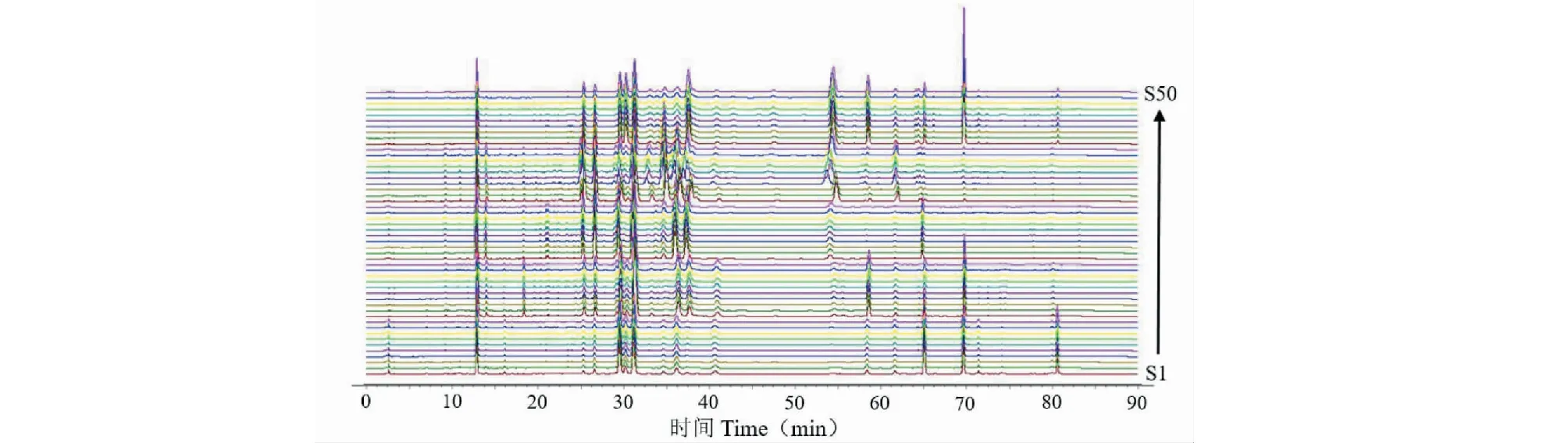
图2 50批菊花样品HPLC叠加色谱图
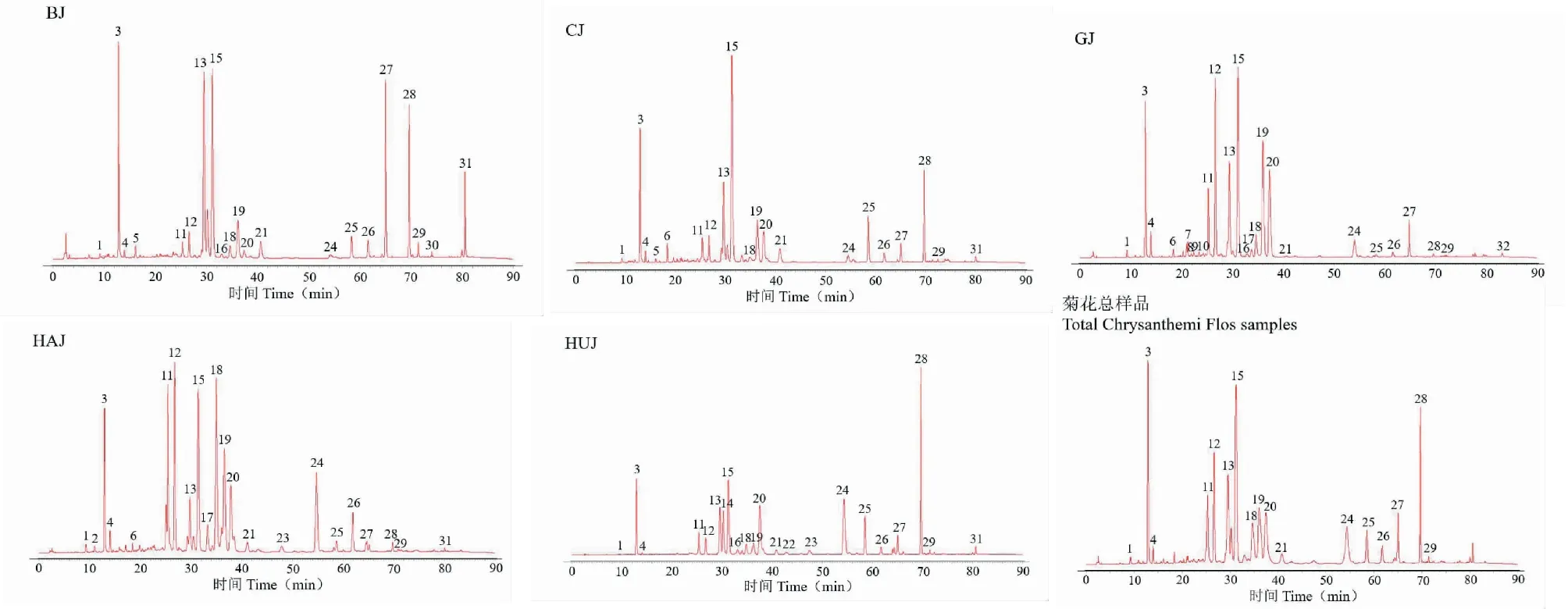
图3 不同品种菊花对照图谱及菊花总样品对照图谱
2.5.2 特征峰的确认
通过分析确定10批亳菊、滁菊、贡菊、杭菊、怀菊的共有峰分别为21、20、25、22、22个;50批菊花样品共有峰有17个。经与混合对照品色谱峰比对,亳菊确认其中14个主要特征峰;滁菊确认其中14个主要特征峰;贡菊确认其中12个主要特征峰;杭菊确认其中13个主要特征峰;怀菊确认其中13个主要特征峰;50批菊花样品确认其中12个共有峰,分别为新绿原酸(1号峰)、绿原酸(3号峰)、隐绿原酸(4号峰)、木犀草素-7-O-β-D-葡萄糖苷(12号峰)、异绿原酸B(13号峰)、3,5-O-二咖啡酰基奎宁酸(15号峰)、芹菜素-7-O-β-D-葡萄糖苷(18号峰)、异绿原酸C(19号峰)、香叶木素-7-O-β-D-葡萄糖苷(21号峰)、香叶木素(26号峰)、芹菜素(28号峰)、木犀草素(29号峰)。在50批菊花药材特征图谱中,3号峰的分离度大、对称性良好、峰面积较大且为所有样品共有,因此选取3号峰(绿原酸)作为参照峰S,计算各共有峰的相对保留时间和相对峰面积。
2.5.3 似度评价
将亳菊、滁菊、贡菊、杭菊、怀菊药材的图谱数据分别导入“中药色谱指纹图谱相似度评价系统(2012.130723版本)”软件,生成相对应的对照图谱,计算各色谱图组内的相似度,结果显示,亳菊之间的相似度为0.992~1.000;滁菊之间的相似度为0.996~1.000;贡菊之间的相似度为0.995~1.000;杭菊之间的相似度为0.971~0.989;怀菊之间的相似度为0.999~1.000,说明同一品种菊花之间差异不大。再将50批菊花药材的图谱数据依次导入该软件,进行组间相似度分析,结果见表2,结果显示种间相似度在0.391~0.690范围内,说明不同品种菊花之间的差异较大。50批菊花药材的17个共有峰相对保留时间的RSD值均小于0.80%,但相对峰面积差异较大,RSD值为1.39%~58.87%。

表2 50批菊花样品的相似度分析
2.5.4 聚类分析(CA)
将50批菊花样品中17个共有峰的峰面积导入SPSS 26.0软件,利用“描述统计”方法标准化后,采用组间联接法,以平方欧式距离为测度进行系统聚类分析,结果见图4。50批菊花样品可被准确地分为5类,即五个品种的菊花各自聚为一类。
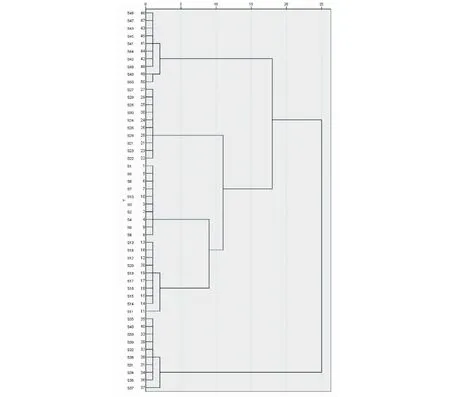
图4 50批菊花样品聚类分析图
2.5.5 主成分分析(PCA)
以主成分的特征值和累积贡献率为依据,17个共有峰峰面积为变量,采用SPSS 26.0软件对50批菊花药材进行PCA分析,相关系数的特征值和方差贡献率见表3。将特征值>1为提取标准,可提取出5个主成分,其累积方差贡献率达98.955%,可以代表菊花药材特征图谱共有峰的大部分信息。为进一步分析50批菊花样品之间的差异,用SIMCA-P 14.1软件绘制50批菊花样品的主成分得分图,见图5。5个不同品种的菊花药材明显地分布于各自的特定区域,其中亳菊、滁菊、怀菊与贡菊、杭菊又明显分布于左右两个区域。位于长江以南的杭菊、贡菊以茶菊为主,兼顾药用;位于长江以北的亳菊、滁菊和位于河南的怀菊则以药用为主,兼顾茶用[1]。根据主成分得分图可以看出不同品种菊花之间有一定的相似性,相同品种菊花之间也存在差异性。怀菊样品间离散度较大;贡菊与杭菊偏于茶饮、同位于长江以南而处于图中右区域;亳菊、滁菊、怀菊因偏于药用而同处于左区域,但由于产地不同,所以怀菊与亳菊、滁菊虽聚为一类但内部差异较明显,说明菊花的品质与产地、品种等因素密切相关。
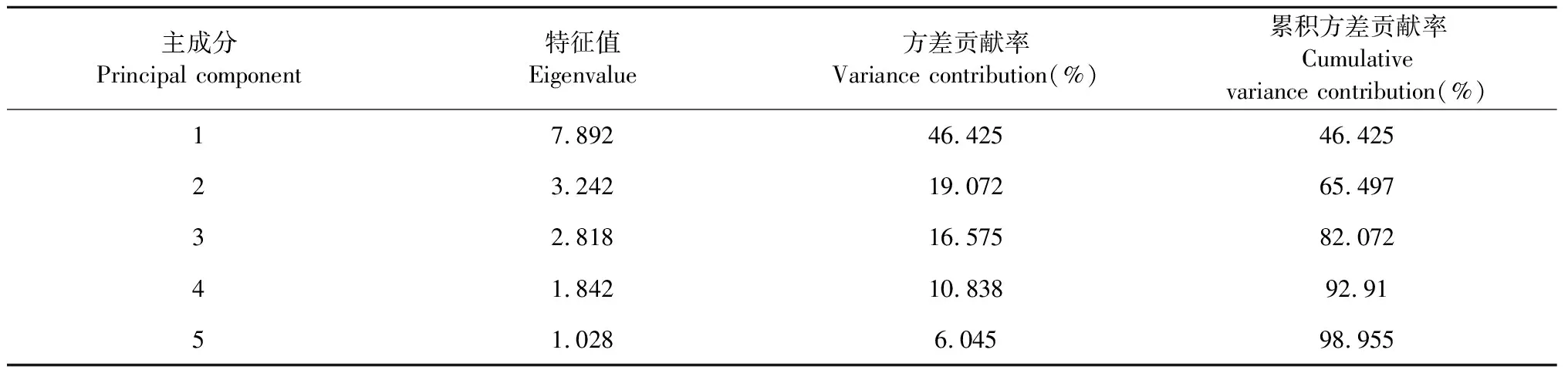
表3 特征值和方差贡献率
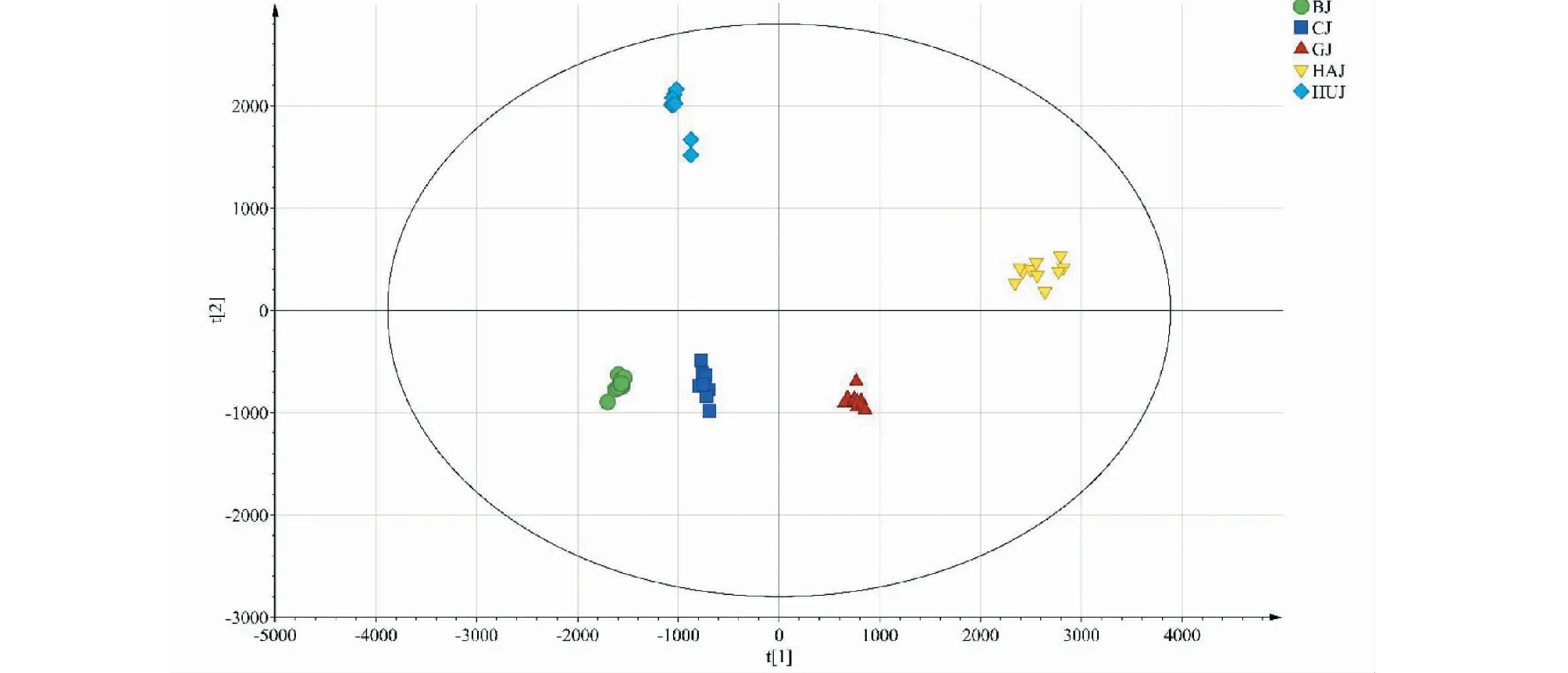
图5 50批菊花样品主成分分析得分图
2.5.6 正交偏最小二乘-判别分析(OPLS-DA)
在主成分分析提取得到的5个主成分的基础上,建立监督模式偏最小二乘判别分析模型,进一步寻找不同品种菊花的差异标志物。在建立的偏最小二乘判别分析模型中,R2X和R2Y分别为0.993和0.991,Q2为0.989,表明建立的模型预测能力较好。为防止模型出现过拟合,利用200次置换检验对模型进行内部验证,检验参数R2=(0.0,0.010 6),Q2=(0.0,-0.384),结果见图6。最右边的R2和Q2值大于0.9,高于左边的R2和Q2值,Q2回归线的截距为负值,表明模型中不存在过拟合现象,结果可靠。偏最小二乘判别分析得分图见图7。50批样品被分成5组,5组之间以左右划分又被分为两大类,Ⅰ类包括贡菊、杭菊,Ⅱ类包括亳菊、滁菊、怀菊。该结果与聚类分析及主成分分析结果一致,说明不同品种的菊花化学成分存在差异。变量重要性投影值(VIP)图见图8,筛选出贡献率较大的7个变量(VIP>1),依次为24、28(芹菜素)、18(芹菜素-7-O-β-D-葡萄糖苷)、12(木犀草素-7-O-β-D-葡萄糖苷)、19(异绿原酸C)、11、20号峰,说明不同品种菊花的化学成分差异可能是由上述成分引起的,这七个成分可作为不同品种菊花之间差异性的主要特征标志物。13(异绿原酸B)、25、15(3,5-O-二咖啡酰基奎宁酸)、27、26(香叶木素)、4(隐绿原酸)、3(绿原酸)、1(新绿原酸)、29(木犀草素)、21(香叶木素-7-O-β-D-葡萄糖苷)号峰的VIP<1,且为5个不同品种菊花所共有,这些成分在判断是否为菊花中起到重要作用,可作为菊花的特征标志物(见表4)。
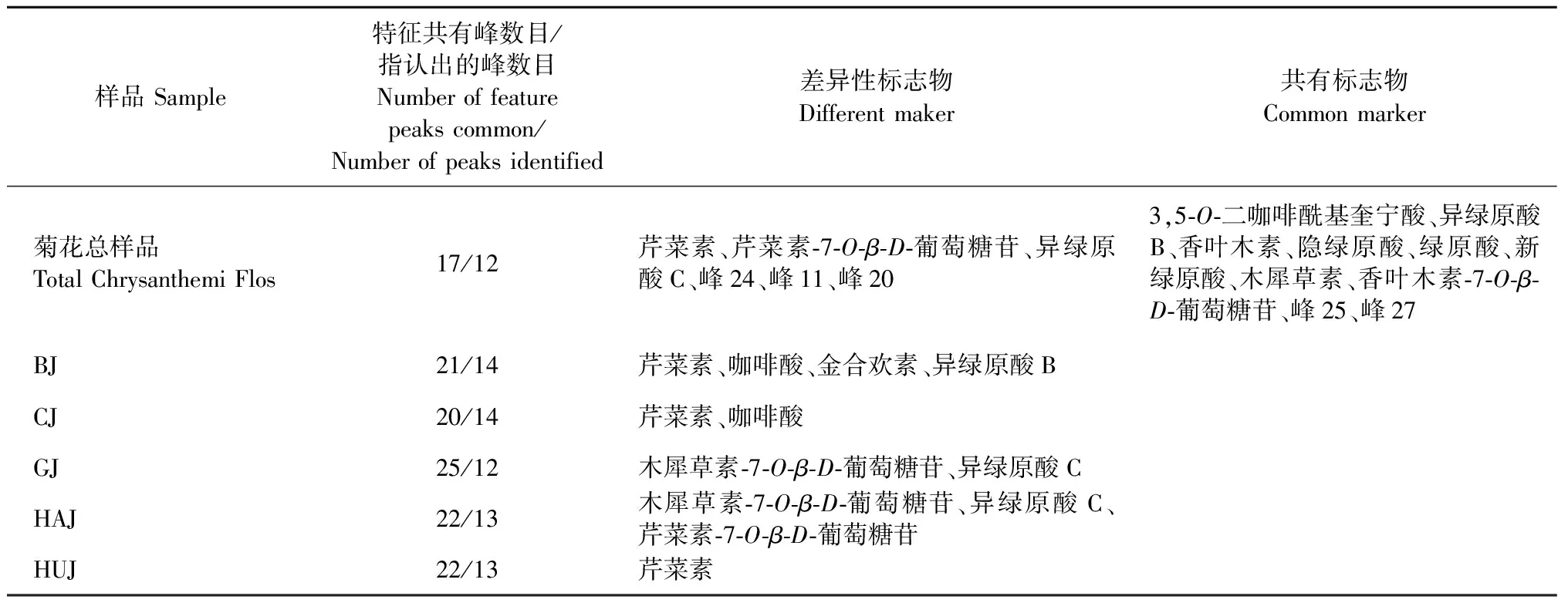
表4 菊花特征标志物

图6 50批菊花PLS-DA模型置换检验图
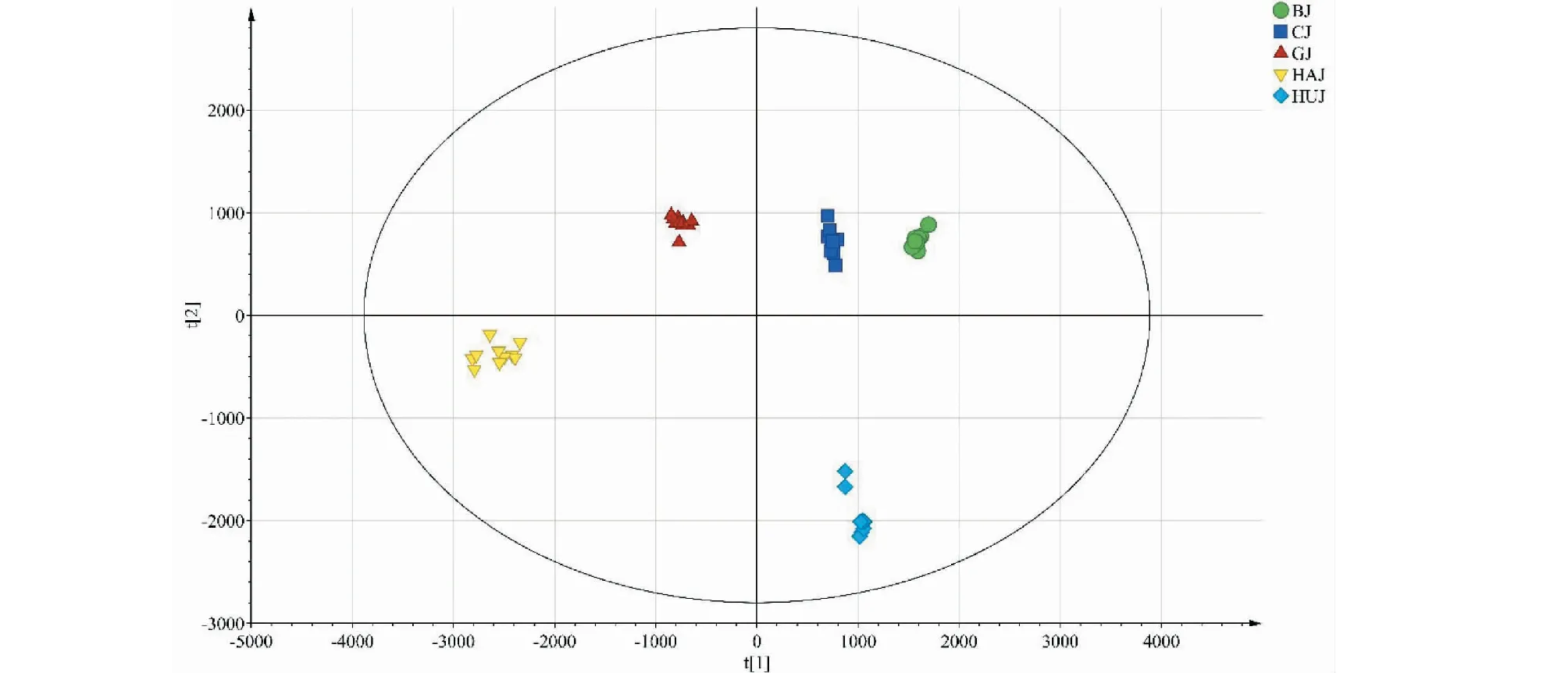
图7 50批菊花样品PLS-DA得分分布图
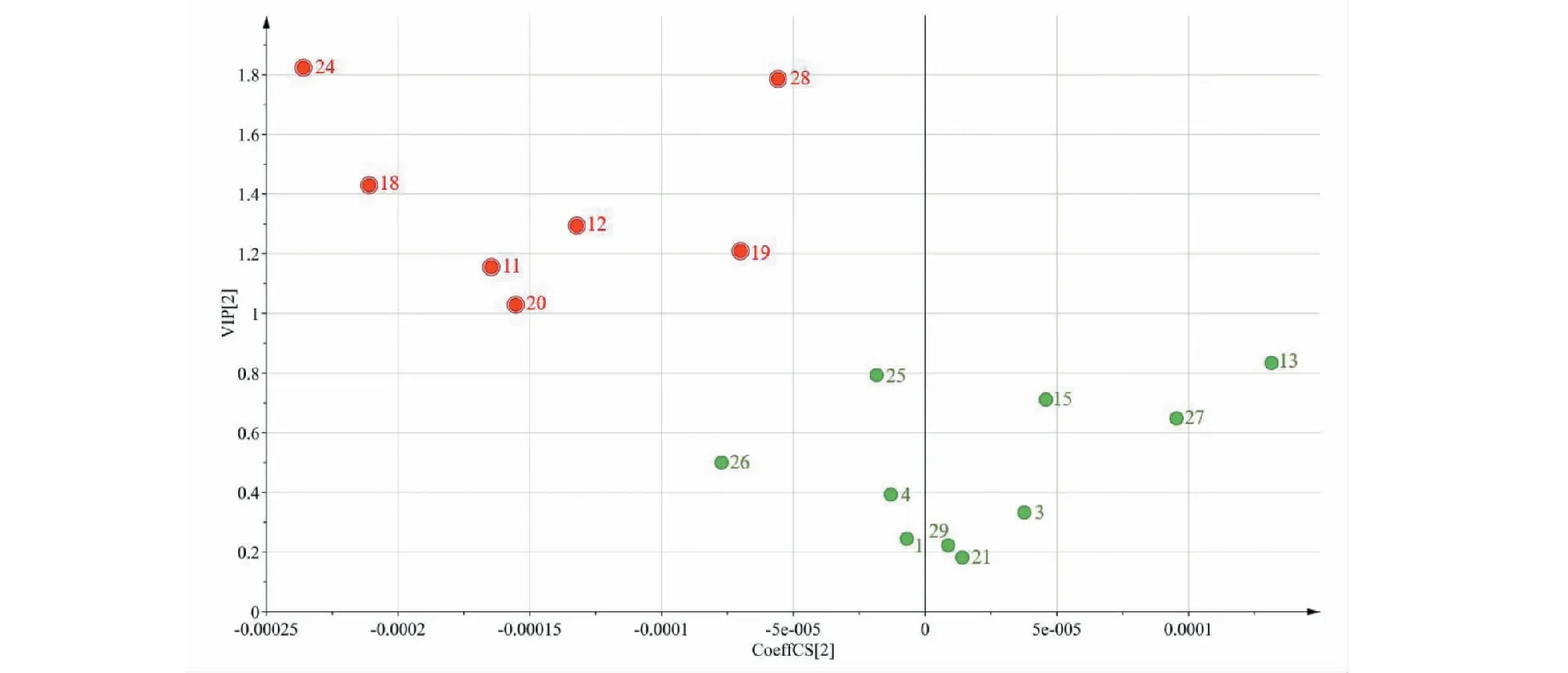
图8 50批菊花样品的PLS-DA载荷图
3 讨论与结论
3.1 提取方法的选择
本实验分别考察了不同溶剂(70%甲醇、50%甲醇、80%甲醇)、提取方式(超声提取和加热回流提取)和提取时间(30、35、40 min)对色谱图的影响,确定采用体积分数为70%甲醇超声提取40 min作为提取方法。
3.2 色谱条件的优化
本实验分别考察[20]了不同的色谱柱(Agilent XDB-C18色谱柱、Phenomenex Luna C18)、检测波长(220、254、330、348 nm)及流动相体系(乙腈-0.1%磷酸水、乙腈-0.1%甲酸水、乙腈-0.2 %醋酸水),并以色谱峰峰形、出峰个数及分离度等为评价指标,最终选用Agilent XDB-C18色谱柱(4.6 mm×250 mm,5 μm),检测波长为348 nm,以乙腈-0.2%醋酸水溶液作为流动相体系。
3.3 结果分析
实验所建立的5个不同品种菊花对照图谱及菊花对照图谱中标定的共有峰和指认的特征峰有所不同,在建立的亳菊、滁菊、贡菊、杭菊、怀菊对照图谱中分别标定21、20、25、22、22个共有峰,并通过与对照品比对指认出14、14、12、13、13个主要特征峰;在50批菊花样品共同生成的菊花对照图谱中标定17个共有峰,指认出新绿原酸、绿原酸、隐绿原酸、木犀草素-7-O-β-D-葡萄糖苷、异绿原酸B、3,5-O-二咖啡酰基奎宁酸、芹菜素-7-O-β-D-葡萄糖苷、异绿原酸C、香叶木素-7-O-β-D-葡萄糖苷、香叶木素、芹菜素、木犀草素12个成分,由于贡菊、杭菊和怀菊中未指认出咖啡酸,贡菊中未指认出金合欢素,因此咖啡酸和金合欢素未作为菊花对照图谱的共有峰。通过对比峰面积,显示木犀草素-7-O-β-D-葡萄糖苷、芹菜素、芹菜素-7-O-β-D-葡萄糖苷和异绿原酸C所对应的峰面积在5个菊花对照图谱之间存在显著性差异:在贡菊和杭菊对照图谱中木犀草素-7-O-β-D-葡萄糖苷、异绿原酸C的峰面积较大,而在其他3种菊花对照图谱中其峰面积均较小;在亳菊、滁菊与怀菊对照图谱中芹菜素的峰面积较大,而在贡菊和杭菊对照图谱中显示其峰面积很小;在杭菊对照图谱中芹菜素-7-O-β-D-葡萄糖苷的峰面积较大,而在其他4种菊花对照图谱中其峰面积较小。所以通过对比5个品种菊花特征峰之间的差异可以初步地反映出不同品种菊花之间的差异性,表明不同品种菊花中化学成分的种类和含量有一定差异。
相似度评价结果显示种内相似度较高,说明来自不同批次的同一品种菊花之间差异较小;而种间相似度低为0.391~0.690,表明不同品种菊花所含的化学成分差异较明显。CA、PCA及OPLS-DA均可将5个不同品种的菊花较为准确地分成5类。5个品种的菊花虽各不相同但也有相似之处,PCA及OPLS-DA将5个特定区域分为左右两大类,其中亳菊、滁菊和怀菊被分为一类(偏药用),贡菊和杭菊被分为一类(偏茶用),化学计量学分析表明木犀草素-7-O-β-D-葡萄糖苷、异绿原酸C、芹菜素为两大类别的主要差别性成分,这3个成分可以作为区分两类菊花差异的特征标志物。OPLS-DA筛选出7个主要标记性成分,通过与混合对照品比对指认出芹菜素、芹菜素-7-O-β-D-葡萄糖苷、木犀草素-7-O-β-D-葡萄糖苷和异绿原酸C 4个成分,可作为区分不同品种菊花差异的特征标志物,与特征峰峰面积对比结果相互佐证,其中24、11、20号色谱峰未指认,这三个色谱峰在菊花质量评价中的作用还有待进一步研究;VIP值小于1的异绿原酸B、3,5-O-二咖啡酰基奎宁酸、香叶木素、隐绿原酸、绿原酸、新绿原酸、木犀草素、香叶木素-7-O-β-D-葡萄糖苷等10个成分可以作为菊花整体的特征标志物。
综上,本实验通过建立并分析5种菊花的HPLC特征图谱,比较特征峰及其对应峰面积,并进行相似度评价,初步显示出不同品种菊花之间的差异性;通过化学计量学方法分析筛选出不同品种菊花之间相同的物质作为代表菊花功效的特征标志物和体现不同菊花差异的特征标志物,为菊花的品种鉴别及质量评价提供科学依据。
