白花蛇舌草化学成分及其抗肿瘤活性研究
2022-09-13梁艳妮侯宝龙吴柯楠吴冬芝
王 婷,梁艳妮,侯宝龙,吴柯楠,吴冬芝,裴 刚,王 征*
1陕西中医药大学 陕西中药资源产业化省部共建协同创新中心 秦药特色资源研究开发国家重点实验室(培育)陕西省创新药物研究中心,咸阳 712083;2湖南中医药大学药学院,长沙 410208
白花蛇舌草为茜草科耳草属植物白花蛇舌草(HedyotisdiffusaWilld)的干燥全草[1],其性寒,味苦、甘淡,归心、肝、脾、大肠经,全草入药,主要分布于我国广西、福建、安徽、云南等地区。研究报道白花蛇舌草含有环烯醚萜类、含酸化合物、蒽醌类化合物、挥发性成份、苯丙素类、香豆素及多糖类等几十种化合物,还有多种微量元素[2],具有抗肿瘤[3]、消炎抗菌[4]、抗氧化[5]、增强免疫力[6]等多种药理活性。在中药抗肿瘤复方治疗中白花蛇舌草为常见药材,其抗肿瘤活性包括抑制肿瘤细胞增殖、诱导肿瘤细胞凋亡和肿瘤血管生成、调节宿主免疫反应以及保护性自噬[7],被广泛应用于消化系统肿瘤、生殖系统肿瘤、呼吸系统肿瘤、骨髓瘤等多种抗癌治疗[8]。白花蛇舌草中主要的抗肿瘤成分为熊果酸和齐墩果酸,熊果酸抗肿瘤机制是通过触发细胞凋亡、细胞周期停滞以及通过各种分子靶点和信号通路发挥抗转移和抗血管生成特性来抑制乳腺癌和结肠癌细胞系的生长[9],齐墩果酸则是通过多个作用靶点同时调控不同信号通路与生物学过程从而诱导细胞凋亡,其中包括细胞内钙离子水平、NF-κB信号通路、Notch信号通路、JAK/STAT3信号通路以及核糖聚合酶的表达等[10]。近年来诸多学者对白花蛇舌草的化学成分及药理作用进行大量研究,针对其不同部位不断分离出新的化合物白花蛇舌草的药效物质基础。截至目前,从白花蛇舌草中已经分离得到了171种化合物,包括32种环烯醚萜类化合物[11]、26种黄酮类化合物[12]、24种蒽醌类化合物[13]、26种酚类化合物及其衍生物[14]和50种挥发油类化合物[15],这些植物化学物质和提取物具有多种药理活性,如抗肿瘤、抗氧化、抗炎、抑制成纤维细胞增殖、调节免疫功能和保护神经系统等作用。本实验将对白花蛇舌草氯仿部位和乙酸乙酯部位进行分离纯化,通过体外细胞实验来检测白花蛇舌草不同极性部位抗肿瘤活性。
1 材料与方法
1.1 材料与仪器
电子分析天平(梅特勒托利多仪器公司);旋转蒸发仪(巩义市予华仪器有限责任公司);电热恒温鼓风干燥箱(上海一恒科技有限公司);循环水式真空泵(巩义市予华仪器有限责任公司);超声波清洗机(宁波新芝生物科技股份有限公司);FL-HO50G型制备型液相(天津博纳艾杰尔科技有限公司);薄层色谱硅胶GF254(青岛海洋化工有限公司);柱色谱硅胶(200~300目、300~400目,青岛海洋化工有限公司);葡聚糖凝胶 Sephadex LH-20(Pharmacia 公司);精密电子天平(METTLER TOLEDO);超净工作台(苏州安泰空气技术有限公司);二氧化碳培养箱(上海新苗医疗器械制造有限公司);倒置显微镜(Motic公司);低速冷冻离心机(美国Thermo公司);超低温冰箱(美菱公司);酶标仪(北京普朗新技术有限公司);甲醇、乙酸乙酯、乙醇、二氯甲烷(均为分析纯,天津恒兴试剂有限公司);氘代三氯甲烷、氘代甲醇(CIL公司);制备型甲醇(美国TEDIA公司);纯净水(华润怡宝饮料有限公司);顺铂(Sigma公司);白花蛇舌草于2017年10月购自于湖南福泰中药饮片有限公司,人肺癌细胞A549 购于中科院干细胞库。
1.2 实验方法
1.2.1 化学成分的提取
取白花蛇舌草全草7 kg粉碎,加入10倍量80%乙醇回流提取2次,每次提取1.5 h,减压浓缩提取液。浓缩后的提取液依次用石油醚、氯仿、乙酸乙酯、正丁醇萃取,各部分萃取液浓缩,回收溶剂,加热干燥,得白花蛇舌草石油醚部位浸膏、氯仿部位浸膏、乙酸乙酯部位浸膏、正丁醇部位浸膏。其中白花蛇舌草氯仿部位浸膏约为86.99 g,乙酸乙酯部位浸膏约为41 g。
1.2.2 白花蛇舌草氯仿部位分离纯化
取白花蛇舌草氯仿部位(86.99 g)进行硅胶(200~300目)柱色谱分离,采用二氯甲烷-甲醇(100∶1→0∶100)梯度洗脱,薄层色谱检测合并相同流分,得到7个不同流分(Fr.1~Fr.7)。Fr.1(1.3 g)经硅胶(200~300目)柱色谱,以石油醚-乙酸乙酯(50∶1→0∶1)为流动相梯度洗脱分离,得到10个流分(Fr.1-A~Fr.1-J)。Fr.1-B浓缩后有白色固体析出,用石油醚和乙酸乙酯多次重结晶得到化合物1(4.8 mg)。Fr.2(9.7 g)经硅胶(200~300目)柱色谱,以石油醚-乙酸乙酯(10∶1→0∶1)为流动相梯度洗脱分离,得到18个流分(Fr.2-A~Fr.2-R)。Fr.2-E通过硅胶(200~300目)柱色谱分离纯化,石油醚-乙酸乙酯(10∶1)为流动相,重结晶得到化合物2(7.3 mg)。Fr.2-H通过硅胶(200~300目)柱色谱分离纯化,石油醚-乙酸乙酯(9∶1)为流动相,重结晶得到化合物3(5.3 mg)。Fr.2-I(90 mg)通过硅胶(200~300目)柱色谱分离纯化,石油醚-乙酸乙酯(4∶1)为流动相梯度洗脱收集流分,得到14个次级流分(Fr.2-I-a—Fr.2-I-n)。Fr.2-I-h(130 mg)经石油醚-乙酸乙酯(32∶1→28∶1)梯度洗脱,再通过Sephadex LH-20,以二氯甲烷-甲醇(1∶1)为流动相进行纯化,得到化合物4(6.7 mg)。Fr.3(20.06 g)经硅胶(200~300目)柱色谱,以二氯甲烷-甲醇(100∶1→0∶100)为流动相梯度洗脱分离,得到10个流分(Fr.3-A~Fr.3-J)。Fr.3-C(7.87 g)经硅胶(200~300目)层析分离,以石油醚-乙酸乙酯(10∶1→0∶1)为洗脱剂,进行梯度洗脱得到20个次级流分(Fr.3-C-a~Fr.3-C-t)。Fr.3-C-e经多次重结晶得到白色粉末,即为化合物5(40 mg)。Fr.3-C-l(0.97 g)通过硅胶(300~400目)层析分离,流动相为石油醚-乙酸乙酯(10∶1)梯度洗脱,得到9个次级流分(Fr.3-C-l-Ⅰ~FFr.3-C-l-Ⅸ)。Fr.3-C-l-Ⅲ分离纯化后析出固体,重结晶得到化合物6(3.5 mg)。Fr.3-C-g通过Sephadex LH-20(二氯甲烷∶甲醇=1∶1洗脱)色谱柱、FL-HO50G制备型液相色谱系统(制备甲醇∶水梯度洗脱,100∶0→85∶15)进行分离纯化,得到化合物7(3.1 mg)。Fr.4(18.56 g)经硅胶(200~300目)层析分离,二氯甲烷-甲醇(80∶1→0∶100)为流动相梯度洗脱,得到14个流分(Fr.4-A~Fr.4.N)。Fr.4-N有白色粉末析出,用氯仿和甲醇多次重结晶得到化合物8(7.2 mg)。
1.2.3 白花蛇舌草乙酸乙酯部位分离纯化
取白花蛇舌草乙酸乙酯部位(41 g)进行硅胶(200~300目)柱色谱分离,以二氯甲烷和甲醇(100∶0→0∶100)为流动相梯度洗脱,得到9个不同流分(Fr.1~Fr.9)。Fr.2(63 mg)经硅胶(300~400目)层析分离,用石油醚和乙酸乙酯(30∶1→0∶1)为流动相梯度洗脱得到12个流分(Fr.2-A~Fr.2-L)。Fr.2-G通过硅胶(300~400目)柱色谱分离纯化,流动相为石油醚-乙酸乙酯(26∶1),重结晶得到化合物9(3.8 mg)。Fr.3(1.32 g)经硅胶(300~400目)层析柱,石油醚和乙酸乙酯(30∶1→0∶1)为洗脱剂,梯度洗脱得到12个流分(Fr.3-A~Fr.3-L)。Fr.3-H通过Sephadex LH-20[二氯甲烷-甲醇(1∶1)洗脱]进行分离纯化,得到化合物10(4.3 mg)。Fr.3-J 通过Sephadex LH-20[二氯甲烷-甲醇(1∶1)洗脱]分离纯化,得到化合物11(5 mg)。Fr.4(5.7 g)用硅胶(300~400目)层析柱,以石油醚和乙酸乙酯(25∶1→0∶1)为洗脱剂进行梯度洗脱,得到12个流分(Fr.4-A—Fr.4-L)。Fr.4-L(2.77 g)通过硅胶(300~400目)层析柱,用石油醚-乙酸乙酯(10∶1→0∶1)为洗脱剂进行梯度洗脱,得到6个次级流分(Fr.4-L-a~Fr.4-L-f)。Fr.4-L-d有明显的黄色固体,经多次重结晶得到化合物12(8.8 mg)。Fr.8(4.68 g)通过硅胶(300~400目)层析柱,用石油醚-乙酸乙酯(1∶0→0∶1)为洗脱剂进行梯度洗脱,得到8个流分(Fr.8-A~Fr.8-H)。Fr.8-E(2.03 g)通过硅胶柱色谱分离纯化,流动相为乙酸乙酯-甲醇(0∶1→0∶1),梯度洗脱得到11个次级流分(Fr.8-E-a~Fr.8-E-k)。Fr.8-E-e通过Sephadex LH-20进行分离纯化,流动相为二氯甲烷-甲醇(1∶1),得到化合物13(3.3 mg)。
1.3 CCK8法检测白花蛇舌草化学成分对肺癌细胞A549增殖作用的影响
取对数生长期的A549细胞,将密度调制成5×104个/mL,接种于96孔板,用含有10%胎牛血清和1%双抗的F-12K培养基于37 ℃、5% CO2培养箱中培养24 h,培养体积为200 μL。把实验分为2组:正常组(细胞+培养基),给药组(加入6个浓度的白花蛇舌草氯仿部位和乙酸乙酯部位提取物溶液,使其在培养液中最终浓度为12.5、25、50、100、200、400 μg/mL,细胞筛选过程是将药物终浓度设定为100 μmol/L,测定IC50实验过程是将浓度设定为1、4、8、12、16、20 μmol/L)。药物刺激24 h后去掉培养基,100 μL PBS清洗1次,而后加入用F-12K培养基新鲜制备的不同浓度的药物100 μL刺激细胞,CCK8 10 μL,继续置入培养箱中培养2 h,取出96孔板,在酶标仪上读取450 nm下的各孔吸光度值,以此反映药物对细胞的毒性作用。根据以下公式计算出细胞抑制率。
细胞抑制率=(A空白-A药物)/A空白×100%
式中:A空白:具有细胞、CCK8溶液而没有药物的吸光度值;A药物:具有细胞、CCK8和药物的吸光度值。
1.4 统计方法

2 实验结果
2.1 结构鉴定
化合物1白色粉末;mp.37~40 ℃;ESI-MS:m/z481[M+H]+;1H NMR(600 MHz,CDCl3)δ:4.08(2H,t,J=7.0 Hz,H-1′),2.31(2H,t,J=6.5 Hz,H-2),1.64(4H,m,H-14,H-16′),1.28(50H,m,H-2~H-12,H-2′~H-15′),0.90(6H,t,J=7.5 Hz,H-15,H-17′);13C NMR(150 MHz,CDCl3)δ:74.3(C-1),34.4(C-2),25.9(C-3),29.2(C-4),29.7(C-5),31.9(C-6),31.9(C-7),31.9(C-8),31.9(C-9),31.9(C-10),31.9(C-11),29.7(C-12),28.6(C-13),22.7(C-14),14.1(C-15),64.5(C-1′),29.3(C-2′),25.0(C-3′),29.7(C-4′),31.9(C-5′),31.9(C-6′),31.9(C-7′),31.9(C-8′),31.9(C-9′),31.9(C-10′),31.9(C-11′),31.9(C-12′),31.9(C-13′),29.7(C-14′),28.6(C-15′),22.7(C-16′),14.1(C-17′),174.3(COO)。以上数据与文献[16]报道基本一致,故鉴定化合物1为十五酸十七酯。
化合物2白色针晶;mp.170~175 ℃;ESI-MS:m/z413[M+H]+;1H NMR(600 MHz,CDCl3)δ:5.35(1H,dd,J=9.8,5.3 Hz,H-6),5.14(1H,d,J=6.6 Hz,H-22),5.03(1H,d,J=6.6 Hz,H-23),3.52(1H,m,H-3),1.01(3H,s,H-19),0.93(6H,d,J=6.9 Hz,H-26,27),0.90(3H,d,J=6.6 Hz,H-29),0.82(3H,d,J=6.8 Hz,H-21),0.68(3H,s,H-18);13C NMR(150 MHz,CDCl3)δ:37.3(C-1),31.7(C-2),71.8(C-3),42.3(C-4),140.8(C-5),121.7(C-6),28.9(C-7),31.9(C-8,C-25),50.2(C-9),36.5(C-10),21.1(C-11,C-26),39.7(C-12),42.2(C-13),56.9(C-14),24.4(C-15),28.3(C-16),56.0(C-17),12.1(C-18),19.4(C-19),40.5(C-20),21.2(C-21),138.3(C-22),129.3(C-23),51.3(C-24),14.1(C-27),25.4(C-28),12.3(C-29)。以上数据与文献[17]报道基本一致,故鉴定化合物2为豆甾醇。
化合物3黄色针状的结晶;mp.280~281 ℃;ESI-MS:m/z269[M+H]+;1H NMR(600 MHz,CDCl3)δ:8.29(2H,m,H-5,8),8.18(1H,d,J=8.6 Hz,H-4),7.79(2H,m,H-6,7),7.68(1H,d,J=7.8 Hz,H-3),4.06(3H,s,OCH3);13C NMR(150 MHz,CDCl3)δ:146.0(C-1),153.8(C-2),126.5(C-3),127.0(C-4),126.8(C-5),133.7(C-6),133.8(C-7),134.5(C-8),182.5(C-9),182.6(C-10),123.5(C-4a),131.4(C-8a),126.7(C-9a),132.9(C-10a),16.4(7-CH3),62.2(8-OCH3)。以上数据与文献[18]报道基本一致,故鉴定化合物3为2-羟基-1-甲氧基-3-甲基蒽醌。
化合物4黄色晶形;mp.>300 ℃;ESI-MS:m/z269[M+H]+;1H NMR(600 MHz,CD3OD)δ:8.06(1H,d,J=8.5 Hz,H-5),8.03(1H,d,J=8.5 Hz,H-4),7.92(1H,br s,H-1),7.68(1H,br d,H-3),7.47(1H,d,J=2.4 Hz,H-8),7.23(1H,dd,J=8.5,2.4 Hz,H-6),2.54(3H,s,H-11);13C NMR(150 MHz,CD3OD)δ:128.0(C-1),146.5(C-2),135.3(C-3),127.9(C-4),130.8(C-5),122.0(C-6),164.3(C-7),113.4(C-8),184.2(C-9),183.5(C-10),21.9(C-11),132.3(C-4a),136.8(C-8a),134.6(C-9a),126.8(C-10a)。以上数据与文献[19]报道基本一致,故鉴定化合物4为7-hydroxytectoquinone。
化合物5白色粉末;Mp.288~293 ℃;ESI-MS:m/z479[M+Na]+;1H NMR(600 MHz,CDCl3)δ:11.96(1H,s,COOH),5.13(1H,s,H-12),4.30(1H,d,J=4.9 Hz,OH),3.34(1H,d,J=6.3 Hz,H-3),2.11(1H,d,J=11.3 Hz,H-18),1.61(2H,m,H-2),1.24(4H,s,H-1,H-7),0.86(1H,d,J=6.2 Hz,H-5),0.82(1H,d,J=6.4 Hz,H-9);13C NMR(150 MHz,CDCl3)δ:36.9(C-1),24.2(C-2),77.3(C-3),38.8(C-4),55.2(C-5),18.4(C-6),33.2(C-7),40.4(C-8),47.5(C-9),28.7(C-10),23.3(C-11),125.0(C-12),138.7(C-13),42.1(C-14),28.0(C-15),23.7(C-16),47.3(C-17),52.8(C-18),38.7(C-19),39.4(C-20),30.6(C-21),36.8(C-22),27.4(C-23),16.5(C-24),15.7(C-25),17.5(C-26),23.7(C-27),178.8(C-28),17.4(C-29),21.5(C-30)。以上数据与文献[20]报道基本一致,故鉴定化合物5为熊果酸。
化合物6白色针状结晶;mp.>300 ℃;ESI-MS:m/z457[M+H]+;1H NMR(600 MHz,CDCl3)δ:5.29(1H,br s,H-12),3.21(1H,m,H-3),1.27(3H,s,H-27),1.22(3H,s,H-26),1.08(3H,s,H-25),1.01(3H,s,H-23),0.97(3H,s,H-24),0.91(3H,s,H-29),0.86(3H,s,H-30);13C NMR(150 MHz,CDCl3)δ:38.4(C-1),27.1(C-2),79.0(C-3),38.7(C-4),55.2(C-5),18.3(C-6),32.6(C-7),39.2(C-8),47.6(C-9),37.0(C-10),22.9(C-11),122.6(C-12),143.5(C-13),41.6(C-14),27.6(C-15),23.4(C-16),46.4(C-17),41.0(C-18),45.8(C-19),30.6(C-20),33.7(C-21),32.4(C-22),28.1(C-23),(C-24),15.3(C-25),17.1(C-26),25.9(C-27),181.7(C-28),33.0(C-29),23.5(C-30)。以上数据与文献[21]报道基本一致,故鉴定化合物6为齐墩果酸。
化合物7黄色粉末;mp.290~295 ℃;ESI-MS:m/z281[M+H]+;1H NMR(600 MHz,CDCl3)δ:13.1(1H,s,H-1),8.35~8.26(1H,m,H-4),7.83(1H,m,H-5),7.76(1H,m,H-6),7.78(1H,d,J=8.2 Hz,H-2),7.54(1H,d,J=8.2 Hz,H-3),2.77(2H,t,J=7.7 Hz,H-11),1.71~1.40(2H,m,H-12),1.29~1.13(2H,m,H-13),0.96(3H,t,J=7.4 Hz,H-14);13C NMR(150 MHz,CDCl3)δ:132.5(C-1),136.4(C-2),134.7(C-3),127.5(C-4),133.1(C-5),119.5(C-6),160.2(C-7),126.9(C-8),189.1(C-9),182.2(C-10),60.8(C-11),31.9(C-12),29.7(C-13),22.7(C-14),133.7(C-4a),134.8(C-8a),134.2(C-9a),126.9(C-10a)。以上数据与文献[22]报道基本一致,故鉴定化合物7为2-butyl-1-hydroxyanthra-9,10-quinone。
化合物8白色粉末;mp.285~290 ℃;ESI-MS:m/z456[M]+;1H NMR(500 MHz,CDCl3)δ:4.97(1H,d,J=2.5 Hz,H-19),4.80(1H,s,H-28),3.56(1H,m,H-3),3.48(1H,t,J=8.2 Hz,H-16),2.74(1H,m,H-17),2.66(1H,m,H-5),2.23(2H,m,H-6),1.84(3H,s,H-29),1.25(3H,s,H-26),1.10(3H,s,H-25),1.08(3H,s,H-24),1.05(3H,s,H-22),0.85(3H,s,H-23);13C NMR(150 MHz,CDCl3)δ:38.7(C-1),27.4(C-2),79.04(C-3),40.75(C-4),55.39(C-5),8.31(C-6),34.3(C-7),38.8(C-8),50.5(C-9),37.2(C-10),20.8(C-11),25.5(C-12),38.4(C-13),42.4(C-14),29.7(C-15),32.1(C-16),56.3(C-17),46.9(C-18),49.32(C-19),150.4(C-20),30.6(C-21),37.0(C-22),28.0(C-23),5.3(C-24),16.1(C-25),16.0(C-26),14.7(C-27),180.9(C-28),109.6(C-29),19.3(C-30)。以上数据与文献[23]报道基本一致,故鉴定化合物8为白桦脂酸。
化合物9黄色粉末;mp.195~197 ℃;ESI-MS:m/z611[M+H]+;1H NMR(600 MHz,DMSO-d6)δ:12.59(5-OH),10.82(7-OH),9.66(4′-OH),9.17(3′-OH),7.54(1H,d,J=6.1 Hz,H-6′),6.82(1H,d,J=7.8 Hz,H-5′),6.38(1H,s,H-8),6.18(1H,s,H-6),5.07(1H,d,J=6.3 Hz,H-1′′),4.52(1H,s,H-1′′′);13C NMR(150 MHz,DMSO-d6)δ:156.8(C-2),133.7(C-3),177.8(C-4),161.6(C-5),99.1(C-6),164.5(C-7),94.0(C-8),157.0(C-9),104.4(C-10),121.4(C-1′),115.5(C-2′),145.1(C-3′),148.8(C-4′),116.7(C-5′),121.6(C-6′),101.6(C-1′′),74.5(C-2′′),76.8(C-3′′),68.6(C-4′′),76.3(C-5′′),67.4(C-6′′),101.1(C-1′′′),70.8(C-2′′′),70.4(C-3′′′),71.0(C-4′′′),68.2(C-5′′′),18.2(C-6′′′)。以上数据与文献[24]报道基本一致,故鉴定化合物9为芦丁。
化合物10黄色粉末;mp.>300 ℃;ESI-MS:m/z303[M+H]+;1H NMR(600 MHz,CDCl3)δ:7.73(1H,d,J=2.1 Hz,H-2′),7.62(1H,dd,J=8.4,2.1 Hz,H-6′),6.87(1H,d,J=9.6 Hz,H-5′),6.37(1H,d,J=2.1 Hz,H-8),6.17(1H,d,J=2.0 Hz,H-6);13C NMR(150 MHz,CDCl3)δ:148.7(C-2),137.2(C-3),177.2(C-4),162.4(C-5),99.2(C-6),165.5(C-7),94.3(C-8),157.0(C-9),104.5(C-10),124.1(C-1′),115.9(C-2′),146.1(C-3′),47.9(C-4′),116.2(C-5′),121.6(C-6′)。以上数据与文献[18]报道基本一致,故鉴定化合物10为槲皮素。
化合物11白色无定形粉末;mp.220~223 ℃;ESI-MS:m/z165[M+H]+;1H NMR(600 MHz,CD3OD)δ:7.58(1H,d,J=15.9 Hz,H-7),7.44(2H,d,J=8.8 Hz,H-2,H-6),6.80(2H,d,J=8.8 Hz,H-3,H-5),6.26(1H,d,J=15.9 Hz,H-8);13C NMR(150 MHz,CD3OD)δ:127.2(C-1),116.7(C-2,6),115.5(C-3,5),161.1(C-4),146.6(C-7),131.0(C-8),171.0(C-9)。以上数据与文献[25]报道基本一致,故鉴定化合物11为反式对羟基肉桂酸。
化合物12白色无定形粉末;mp.175~180 ℃;ESI-MS:m/z179[M+H]+;1H NMR(600 MHz,CDCl3)δ:7.78(1H,d,J=15.9 Hz,H-7),7.54(2H,d,J=8.7 Hz,H-2,H-6),6.94(2H,d,J=8.7 Hz,H-3,H-5),6.36(1H,d,J=15.9 Hz,H-8),3.52(3H,s,OCH3);13C NMR(150 MHz,CDCl3)δ:126.9(C-1),114.7(C-2,6),114.5(C-3,5),161.8(C-4),146.8(C-7),130.2(C-8),172.1(C-9),55.5(4-OCH3)。以上数据与文献报道数据[26]相符,鉴定化合物12为反式对甲氧基肉桂酸。
化合物13黄色粉末;mp.280~282 ℃;ESI-MS:m/z287[M+H]+;1H NMR(600 MHz,CDCl3)δ:8.08(2H,d,J=9.0 Hz,H-2′,H-6′),6.90(2H,d,J=9.0 Hz,H-3′,H-5′),6.39(1H,d,J=2.1 Hz,H-8),6.17(1H,d,J=2.1 Hz,H-6);13C NMR(150 MHz,CDCl3)δ:148.0(C-2),137.1(C-3),177.4(C-4),162.5(C-5),99.2(C-6),165.6(C-7),94.4(C-8),158.3(C-9),104.5(C-10),123.7(C-1′),115.9(C-2′),116.3(C-3′,5′),160.6(C-4′),130.6(C-6′)。以上数据与文献报道数据[27]相符,鉴定化合物13为山奈酚。
化合物1~13结构见图1。
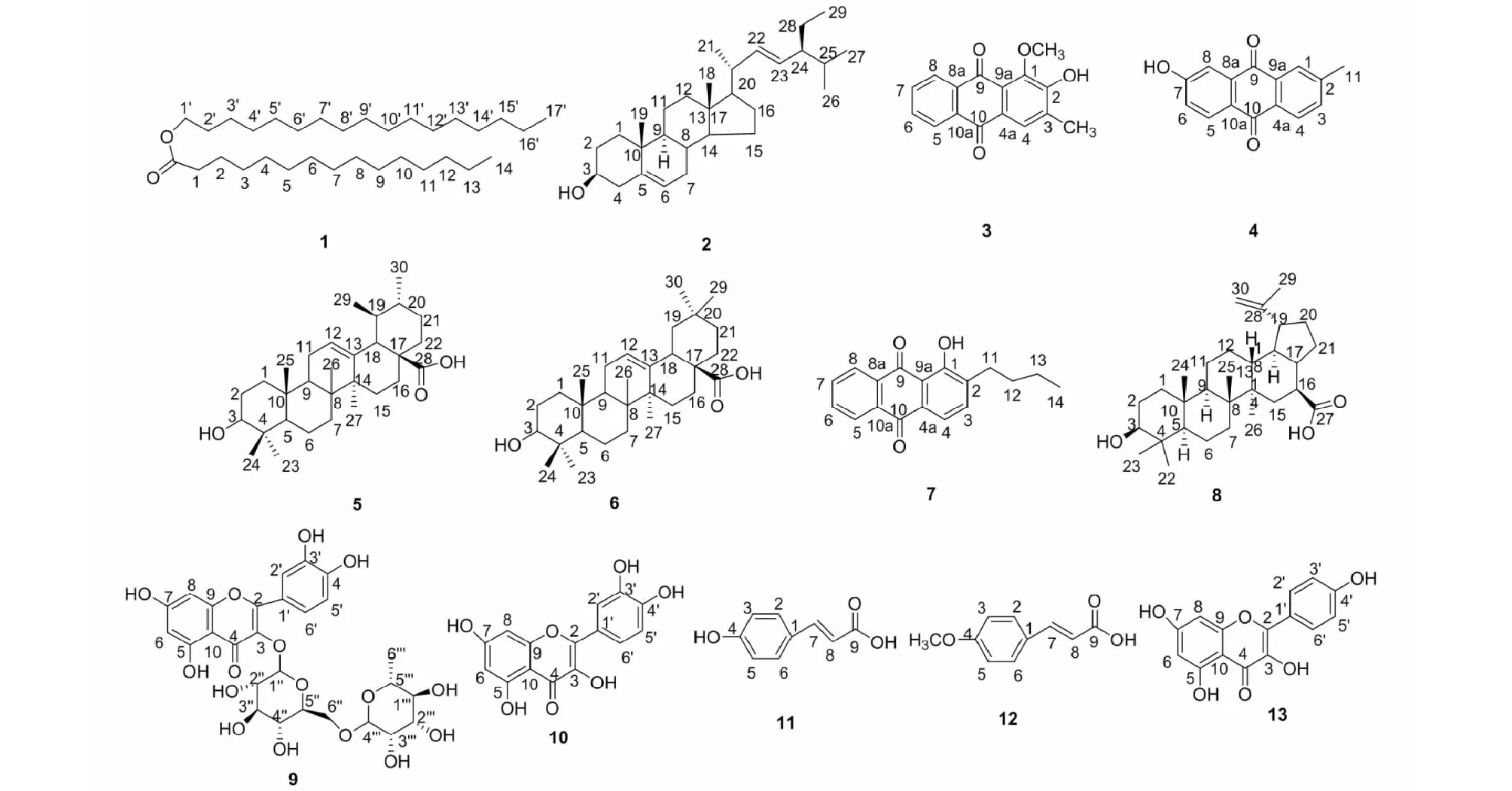
图1 化合物1~13的结构
2.2 体外抗肿瘤活性测试
2.2.1 白花蛇舌草不同极性部位对A549细胞增殖抑制作用
给药作用24 h后,采用CCK8法检测白花蛇舌草氯仿部位及乙酸乙酯部位对A549细胞增殖的影响(顺铂为阳性药对照)结果见表1。结果表明,氯仿部位及乙酸乙酯部位均对A549细胞有明显的抑制作用,且存在量效关系,其IC50值分别为150.73、243.70 μg/mL。当药物浓度从25 μmol/L升至400 μmol/L,氯仿部位和乙酸乙酯部位对A549的细胞增殖抑制率分别从12.9%、43.32%达到80.49%、63.74%。

表1 白花蛇舌草不同极性部位对A549细胞增殖抑制率
2.2.2 白花蛇舌草单体化合物对A549细胞增殖抑制作用
2.2.2.1 单体化合物对A549细胞抑制增殖活性筛选
由表2结果可以看出给药作用24 h后,熊果酸、齐墩果酸、白桦脂酸对A549细胞有较明显的抑制作用,槲皮素对A549细胞抑制作用较弱,而其他化合物对A549细胞无明显的抑制作用。
表2 化合物对A549细胞的增殖抑制率
2.2.2.2 单体化合物对A549细胞增殖的作用
实验结果表明,白桦脂酸、熊果酸、齐墩果酸和顺铂对A549细胞显示出较强的细胞毒性,且存在一定的量效关系,其IC50值分别为13.85、12.81、8.38 μmol/L。而阳性药顺铂对A549细胞的IC50值为17.34 μmol/L,结果见表3。
表3 单体化合物对A549细胞增殖抑制率
3 讨论与结论
本实验对白花蛇舌草氯仿部位和乙酸乙酯部位中的化学成分进行了较为系统地分离,得到了13个化合物,其中化合物十五酸十七酯、7-hydroxytectoquinone、2-butyl-1-hydroxyanthra-9,10-quinone首次从该植物中分离得到。通过CCK8实验检测了白花蛇舌草不同极性部位和单体化合物对A549细胞增殖抑制作用,结果表明,白花蛇舌草氯仿部位和乙酸乙酯部位都有很好的抑制A549细胞增殖的作用,且有明显的剂量-效应关系。白花蛇舌草不同极性部位对A549细胞增殖的影响与顺铂相比,当药物浓度达到200 μg/mL时,仅氯仿萃取物效果优于顺铂,当药物浓度达到400 μg/mL时,氯仿萃取物和乙酸乙酯萃取物的效果均优于顺铂。由此可以确定白花蛇舌草氯仿和乙酸乙酯部位是抗肺癌A549细胞的活性部位,这两个部位将是分离活性物质的目标。通过化合物对A549细胞抑制增殖活性筛选实验,发现白桦脂酸、齐墩果酸和熊果酸在同样浓度下对A549细胞有很好的抑制作用。白桦脂酸和齐墩果酸随着药物浓度增加,抑制率显著增大,达到16~20 μmol/L时都趋于稳定。熊果酸在1~8 μmol/L范围内抑制率较平稳,而后随着浓度增加抑制率增大,达到20 μmol/L时抑制率达到68.45%。在1~20 μmol/L浓度范围内,与阳性对照物顺铂相比,白桦脂酸在浓度为16 μmol/L和20 μmol/L时的抑制率要高于顺铂;熊果酸的浓度超过16 μmol/L后抑制率要高于顺铂;齐墩果酸在4~12 μmol/L时抑制率跟顺铂相当,超过12 μmol/L时抑制率要高于顺铂。在以往的研究中发现白花蛇舌草中萜类、蒽醌类、甾醇类、黄酮类、多糖类等化学成分抗癌作用较为突出,抗肿瘤治疗效果与机制研究多采用白花蛇舌草水提取或醇提物作用于不同肿瘤细胞来观察体外抗肿瘤作用[28]。本实验结果为白花蛇舌草抗肿瘤活性物质基础研究提供理论依据。
