超高效液相色谱-四极杆线性离子阱串联质谱法测定他达拉非原料药中潜在基因毒性杂质的含量
2022-09-13刘家兴李丽美杨庆云臧清策吴松张瑞萍再帕尔阿不力孜中国医学科学院北京协和医学院药物研究所天然药物活性物质与功能国家重点实验室北京00050中央民族大学生物成像与系统生物学研究中心北京0008
刘家兴,李丽美,杨庆云,臧清策,吴松,张瑞萍*,再帕尔·阿不力孜,2(.中国医学科学院/北京协和医学院药物研究所 天然药物活性物质与功能国家重点实验室,北京 00050;2.中央民族大学 生物成像与系统生物学研究中心,北京 0008)
他达拉非(tadalafil,TDF),化学名:6-(1,3-苯间二氧戊环-5-基)-2,3,6,7,12,12a-六氢化-2 甲基-(6R,12aR)-吡嗪并[1'2':1,6]吡啶并[3,4-b]吲哚-1,4-二酮,可以特异性长效抑制磷酸二酯酶5(PDE5),是一种非激素类抗男性勃起功能障碍(ED)药,与其他治疗ED的药物相比,该药具有活性高、不良反应少、半衰期长等优点。他达拉非于2003年11月在美国上市,2005年5月在中国上市。他达拉非的化合物专利ZL95192078.2 已于2015年1月到期,所以研究突破专利的新工艺对于他达拉非仿制药在中国上市具有重要价值,迫切需要开展他达拉非仿制药的质量控制方法研究。
文献报道的他达拉非的合成路线主要有7条,其工业化生产路线是以D
-色氨酸甲酯盐酸盐为起始原料,与胡椒醛进行Pictet-Spengler反应后再经酰化反应、亲核取代及关环反应制得他达拉非(见图1)。其中,(1R
,3R
)-1-(1,3-苯并二氧戊环-5-基)-2-(2-氯乙酰基)-1,2,3,4-四氢-9H-吡啶并[3,4-b]吲哚-3-羧酸甲酯(T2),是基于该路线合成他达拉非过程产生的中间体物质,作为合成他达拉非最后一步反应的原料,不可避免会作为杂质存在于终产品中。
图1 他达拉非工业合成路线Fig 1 Industrial synthetic route of tadalafil
该杂质结构中含有羰基取代的卤代烷基,该基团不仅具有强吸电子特性,还具有强电负性,会产生具有强亲电性的碳原子,易与DNA 等生物大分子发生烷基化反应,被纳入欧盟发布的具有基因毒性的警示结构。本课题组采用欧盟委员会的欧洲化学品局联合研究中心(Joint Research Centre of European Chemicals Bureau)开发的免费毒性预测平台Toxtree(http://toxtree.sourceforge.net/)进一步预测T2 的致癌性和遗传毒性,评价结果为阳性,按照2020年版《中国药典》四部通则9306 遗传毒性杂质控制指导原则,T2 属于第3 类杂质,需要进行毒理学关注阈值限度控制以及进一步的毒理学实验评价。本研究旨在建立一种基于UPLC-q/LIT-MS 技术的专属性强、灵敏度高的他达拉非原料药中潜在基因毒性杂质T2 的定量测定方法。
1 仪器与试药
分析天平(XPE205,Mettler Toledo,USA);ACQUITY UPLC 超高效液相色谱仪(Waters,USA);四极杆-线性离子阱串联质谱仪(QTRAP 5500,AB SCIEX,USA);四极杆-飞行时间串联质谱仪(Triple TOF 5600 +,AB SCIEX,USA);涡旋混合器(MS3,IKA);单通道微量移液器(Eppendorf,USA);Captiva 96 孔板过滤装置(Agilent,USA);杂质对照品(自制);他达拉非原料药(批号分别为TDLF01、TDLF02,自制);甲醇、乙腈(LC-MS 纯,Fisher);乙酸铵(分析纯,Sigma);其余试剂为分析纯;水为娃哈哈纯净水。
2 方法与结果
2.1 分析条件
2.1.1 色谱条件 采用ACQUITY UPLC HSS T3柱(2.1 mm×100 mm,1.8 µm);流动相A 为0.02 mol·L乙酸铵溶液(含0.1%乙酸),流动相B 为乙腈,梯度洗脱(0 ~1 min,70%→21%A;1 ~6.5 min,21%→20%A;6.5 ~8 min,20%→70%A);流速:0.35 mL·min;柱温:25 ℃;进样量:5 µL。
2.1.2 质谱条件 四极杆-线性离子阱串联质谱仪、四极杆-飞行时间串联质谱仪,配有TurboIonspray(ESI)离子源及Analyst1.5.1 分析软件。所使用的各种气路均为氮气。离子源温度500℃;离子源喷射电压5.0 kV;雾化气压力50 psi;辅助气压力50 psi;气帘气压力25 psi。定量分析采用Multiquant 3.0.3 软件进行数据处理。
2.1.3 目标离子的选择 采用Triple TOF 5600 +质谱仪正离子检测模式在m/z
100 ~500 内全扫描,采集得到的杂质中间体的MS质谱图如图2所示。采用QTRAP 5500 质谱仪的MRM 模式进行定量数据采集,用于杂质定量测定的离子对为m/z
427.1 →m/z
135.0,去簇电压为80 V,碰撞能量为30 eV。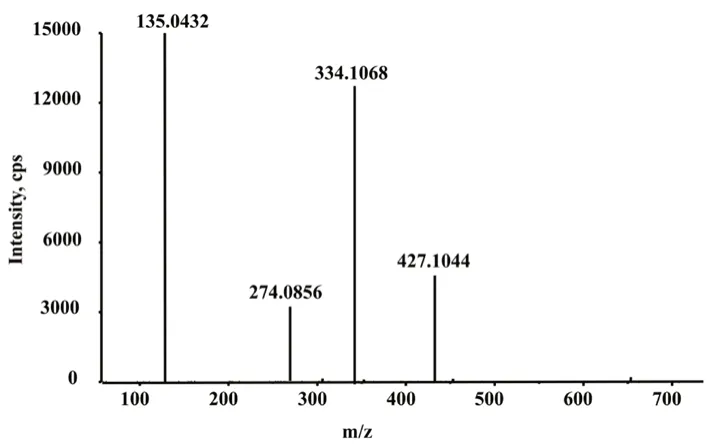
图2 杂质中间体的MS/MS 质谱图Fig 2 MS/MS spectrum of the genotoxic impurity
2.2 溶液制备
2.2.1 杂质对照品储备溶液配制 精密称取杂质对照品适量于量瓶中,加乙腈超声溶解并定容,摇匀,配制成1 mg·mL的杂质对照品储备液。
2.2.2 原料药供试品溶液配制 分别精密称取他达拉非原料药TDLF01 供试品3 份,每份10 mg,置于10 mL 量瓶中,加乙腈超声溶解,并定容至刻度,摇匀,配制成1 mg·mL的原料药供试品储备液一。
分别精密称取他达拉非原料药TDLF02 供试品3 份,每份5 mg,置于5 mL 量瓶中,加乙腈超声溶解,并定容至刻度,摇匀,配制成1 mg·mL的原料药供试品储备液二。
分别稀释上述原料药供试品储备液至含他达拉非5.00 μg·mL,作为杂质含量测定的原料药供试品溶液。
2.3 方法学考察
2.3.1 专属性考察 分别将空白溶剂、杂质对照品储备溶液、原料药供试品溶液进样测定,得到XIC色谱图如图3 所示。杂质中间体的保留时间在3.08 min 左右,空白溶剂与待测样品中均没有干扰杂质中间体含量测定的物质存在,专属性良好。
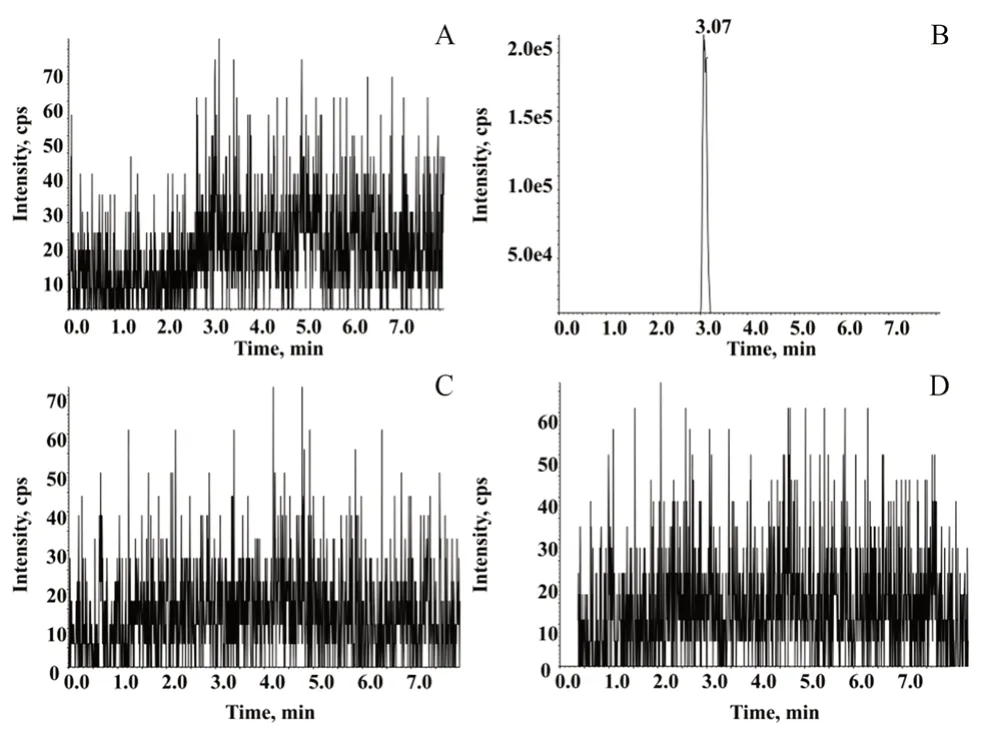
图3 杂质T2 的XIC 色谱图Fig 3 XICs of the genotoxic impurity
2.3.2 线性试验 精密量取杂质对照品储备液,用乙腈稀释制成0.10、0.50、0.80、1.0、5.0、8.0、10、50和80 ng·mL系列对照品溶液,依次进样。以峰面积为纵坐标,质量浓度为横坐标,绘制标准曲线,得回归方程Y
=9.25×10X
+238(r
=0.9996)。结果表明,中间体化合物在0.10 ~80 ng·mL内与峰面积呈良好的线性关系。2.3.3 检测限与定量限 取0.10 ng·mL的对照品溶液,逐级稀释,依次进样,以S/N
=3 时的浓度为检测限,检测限为25 pg·mL,S/N
=10 时的浓度为定量限,定量限为80 pg·mL。2.3.4 精密度
① 日内精密度:精密稀释“2.2.1”项下杂质对照品储备液,用乙腈稀释至0.50、5.0、50 ng·mL3 个质量浓度,平行制备5 份样品,每个样品一日测定3 次,结果其峰面积RSD
分别为3.1%、5.0%、3.4%,结果说明日内精密度良好。② 日间精密度:精密稀释“2.2.1”项下杂质对照品储备液,用乙腈稀释至0.50、5.0、50 ng·mL3 个质量浓度,平行制备5 份样品,连续测定3 d,结果其峰面积RSD
分别为8.2%、7.4%、3.6%,结果说明日间精密度良好。2.3.5 稳定性
① 4℃放置稳定性:精密稀释“2.2.1”项下杂质对照品储备液,用乙腈稀释至5.00 ng·mL,在4℃下放置0、2、4、6、8 h,进样测定,结果峰面积RSD
为4.3%;取“2.2.1”项下的原料药供试品溶液,重复上述操作,结果峰面积RSD
为5.9%,说明溶液在4 ℃放置8 h 内基本稳定。② 室温放置稳定性:精密稀释“2.2.1”项下杂质对照品储备液,用乙腈稀释至5.00 ng·mL,在室温下放置0、12、24 h,进样测定,结果峰面积RSD
为7.1%;取“2.2.2”项下原料药供试品溶液,重复上述操作,结果峰面积RSD
为5.2%,说明溶液室温放置24 h 内基本稳定。2.3.6 加样回收试验 精密称取“2.2.2”项下原料药9 份,每份1.00 mg,置于2 mL 量瓶中,精密加入0.12、0.15、0.18 mL 2.00 μg·mL杂质对照品溶液,用乙腈超声溶解并定容至刻度,摇匀,每个样品平行制备3 份。分别稀释上述溶液至含他达拉非5.00 μg·mL,进样测定,获得相应的杂质峰面积,计算回收率及对应RSD
,结果显示,加样回收率均在90%~110%,RSD
均小于10%,表明方法加样回收率满足要求。2.3.7 耐用性考察 对分析条件进行细微调整,考察了柱温23℃与28℃,流速0.3 mL·min与0.4 mL·min的影响,发现杂质的XIC 图中,色谱峰形无明显变化,保留时间及峰面积发生微小改变,RSD
值均小于5%,表明分析条件的微小变动不影响T2 杂质的检测,方法的耐用性良好。2.4 杂质含量测定
按“2.2.2”项下方法操作,精密配制2 个批次的原料药供试品溶液,每个批次平行制备3 份,按“2.1”项下方法依次测定,结果其含量均低于检测限。
3 讨论
基因毒性杂质因其能对DNA 或RNA 中嘌呤、嘧啶碱基或者磷酸双酯骨架上的N 原子或O 原子等进行亲电进攻,从而造成DNA 双链断裂,空间构象改变等结构异常,进而产生诱导突变、致癌等毒性而受到广泛关注。他达拉非合成工艺杂质(T2)具有基因毒性的警示结构,存在潜在的致癌风险,为保障用药安全,有必要对该杂质进行严格的限量控制。毒理学关注阈值(TTC)的提出进一步规范了对于基因毒性杂质的监管。2006年1月由辉瑞、强生、葛兰素史克等国际制药企业中的研发人员组成的美国药物研究和制造商协会(Pharmaceutical Research and Manufacturers of America,PhRM)专家小组在Lutz Müller 等的领导下首次提出了“阶段化TTC”的概念,即根据不同的暴露时间,设定不同的控制限度,对于有些基因毒性基团,如黄曲霉素类、偶氮苯类、N
-亚硝基物类因具有较高的致癌风险,需要进行特殊的限度控制。人用药物注册技术要求国际协调会(ICH)基因毒性杂质研究指南M7(R1)对于基因毒性杂质监管要求较为宽松,这可以为根据他达拉非的用药剂量和用药周期制订合理的杂质控制标准提供参考。4 结论
本研究建立了他达拉非原料药中具有潜在基因毒性杂质T2 的定量分析方法。该方法简单、快速、回收率好、灵敏度高,填补了国内该方面研究的空白,有助于对他达拉非仿制药合成过程中的质量参数进行有效监测。根据《基因毒性杂质限度指南》,基因毒性杂质限度水平应该小于1.50 µg·d,结合本制剂的用法用量,按每日服用最大量20 mg 计算,以本方法分析时原料药中杂质含量相当于375 pg·mL。本方法的定量限低于该含量水平,因此适用于他达拉非潜在基因毒性杂质T2 的检测,可以满足高于TTC 要求杂质限度的准确分析。
