叉头框转录因子O1在肾纤维化中的作用及其调控
2022-09-08徐仁凤王正朝张正红
徐仁凤, 王正朝, 张正红
(福建师范大学生命科学学院,福建省发育与神经生物学重点实验室,运动与健康福建省高校重点实验室,教育部光电医学重点实验室, 福州 350007)
慢性肾病(chronic kidney disease,CKD)已成为全球不可忽视的一种慢性炎症性疾病。慢性肾病在其初期无明显临床症状,不易被诊断;当被诊断时,病人通常已处于肾病晚期。大量数据表明,肾纤维化是驱动慢性肾病步向终末期肾功能衰竭的共同途径和关键性病理特征[1]。肾纤维化的许多病理学原理与其他纤维化疾病相同,例如肝硬化、心肌病和肺纤维化。据估计,在发达国家,因纤维性疾病而死亡的人数总计占死亡人数的1/2[2]。如果能够减轻甚至逆转肾纤维化的过程,就能改善慢性肾病患者的预后。目前的研究表明,叉头框转录因子(forkhead box transcription factor,Fox)家族O亚族1(FoxO1)与肾纤维化密切相关,可以抑制肌成纤维细胞(myofibroblast,MF)激活与随后细胞外基质(extracellular matrix,ECM)的产生[3-5]。
由此可见,目前的研究已经明确了FoxO1在肾纤维化中的作用及其可能的调控机制。有关FoxO1的深入研究将是肾纤维化发生机制研究的新切入点和新思路,将为慢性肾病的治疗带来一个新的前景。
1 叉头框转录因子O1
Fox是一个蛋白质超家族,该家族目前已发现100多个成员。其中,FoxO是Fox蛋白质家族中的1个亚族,在哺乳动物中由4个成员组成,即FoxO1、FoxO3、FoxO4和FoxO6,它们共享一个高度保守的DNA结合结构域,即叉头结合域[6]。而FoxO1是FoxO家族中最具代表性的成员,由FKHR基因编码,包含4个功能域,分别是核定位信号域(nuclear localization signal domain,NLS)、N-端的核输出信号域(nuclear export signal domain,NES)、转录激活域(transactivation domain,TA)和叉头DNA结合域(forkhead domain,FKH)[7]。其中,FKH是最重要的功能域。在该结构域及其附近,存在3个高度保守的PI-3K/Akt磷酸化位点,分别为Thr24、Ser256与Ser3193。
FoxO1作为人体最重要的转录因子之一,在多种细胞类型中广泛表达,可通过转录和翻译,在细胞增殖、自噬、萎缩、凋亡、代谢、以及炎症反应和氧化应激反应等多种病理生理过程中响应细胞刺激并维持稳态,充分发挥了其作用[7-11]。在子宫内膜中,FoxO1已被证明能在保护怀孕母体妊娠期免受氧化损伤中发挥重要作用[12],同时也参与月经周期和早孕期间子宫内膜的有效重塑[13];抗糖尿病药物AM251通过抑制FoxO1来降低PEPCK(phosphoenolpyruvate carboxykinase,磷酸烯醇丙酮酸羧激酶)和G6酶(glucose 6-phosphatase,葡萄糖6磷酸酶)表达,控制肝中的血糖水平[14];FoxO1激活剂姜黄素能通过上调p27和p21并下调细胞周期蛋白D1(cyclin D1)来减少肺癌的增殖[15]。由于其广泛且复杂的内源性表达和功能,FoxO1可能是预防和治疗疾病的重要靶点。
2 叉头框转录因子O1的功能调控
FoxO1的功能依赖于调控其下游靶基因,包括凋亡和自噬相关基因、抗氧化应激酶、细胞周期阻滞基因以及生长因子和免疫调节因子等[9]。不同靶点的表达与FoxO1的活性一致,直接或间接地诱导各种生理和病理变化。例如,在正常和病理条件下,FoxO1通过调节过氧化氢酶和MnSOD(manganese superoxide dismutase,锰过氧化物歧化酶)等特定抗氧化酶的基因表达介导对细胞的保护作用,这些酶主要是通过催化ROS(reactive oxygen species,活性氧自由基)的转化来保护细胞免受氧化应激损伤[16,17]。然而,当FoxO1位于细胞质中而不在细胞核中时,它会被隔离,并且不能再控制其下游的靶点。
此外,FoxO1的活性主要受其上游的多种信号分子和其转录后修饰调控。位于其上游的信号分子,包括PI-3K、Akt/PKB(protein kinase B,蛋白激酶B)、JNK(c-Jun N-terminal kinase,c-Jun氨基末端激酶)、CBP(CREB-binding protein,CREB结合蛋白)、MAPK、Sirtuins、泛素E3连接酶和SIRT1(silent information regulator 1,沉默信息调节因子1)等,在氧化应激和高糖等刺激下,它们会被激活或抑制,进而调控随后的转录后修饰[18]。其转录后修饰则主要包括磷酸化、泛素化、乙酰化、精氨酸甲基化和O-糖基化等,这些修饰主要是通过修饰FoxO1和14-3-3蛋白的特定位点,并通过影响它们的相互作用,以及FoxO1在细胞核与细胞质间的易位来调节FoxO1的活性[18-20]。磷酸化是FoxO1活性调控最常见的机制,而PI-3K/Akt通路磷酸化FoxO1是导致FoxO1失活的典型过程。高糖条件下,PI-3K/Akt通路被激活,上游激酶PI-3K的磷酸化进一步磷酸化Akt,Akt则从细胞质转运到细胞核,使FoxO1的3个苏氨酸位点磷酸化。FoxO1磷酸化促进了其与14-3-3的相互作用,导致FoxO1从细胞核转位到细胞质,并降低了它们的转录活性,以及在细胞质中通过蛋白酶体诱导其降解来抑制FoxO1的活性[21,22]。
3 肾纤维化
肾纤维化(renal fibrosis)是肾在多种病因,例如发生炎症反应或急性损伤的刺激下自身修复机制被激活后修复过度的结果,是肾组织衰老和慢性肾炎的标志,其主要特征是MF持续活化、ECM过度产生和沉积,即进行性实质破坏,进而导致组织功能逐渐丧失和瘢痕组织形成,最终导致肾功能衰竭[23-26](Fig.1)。
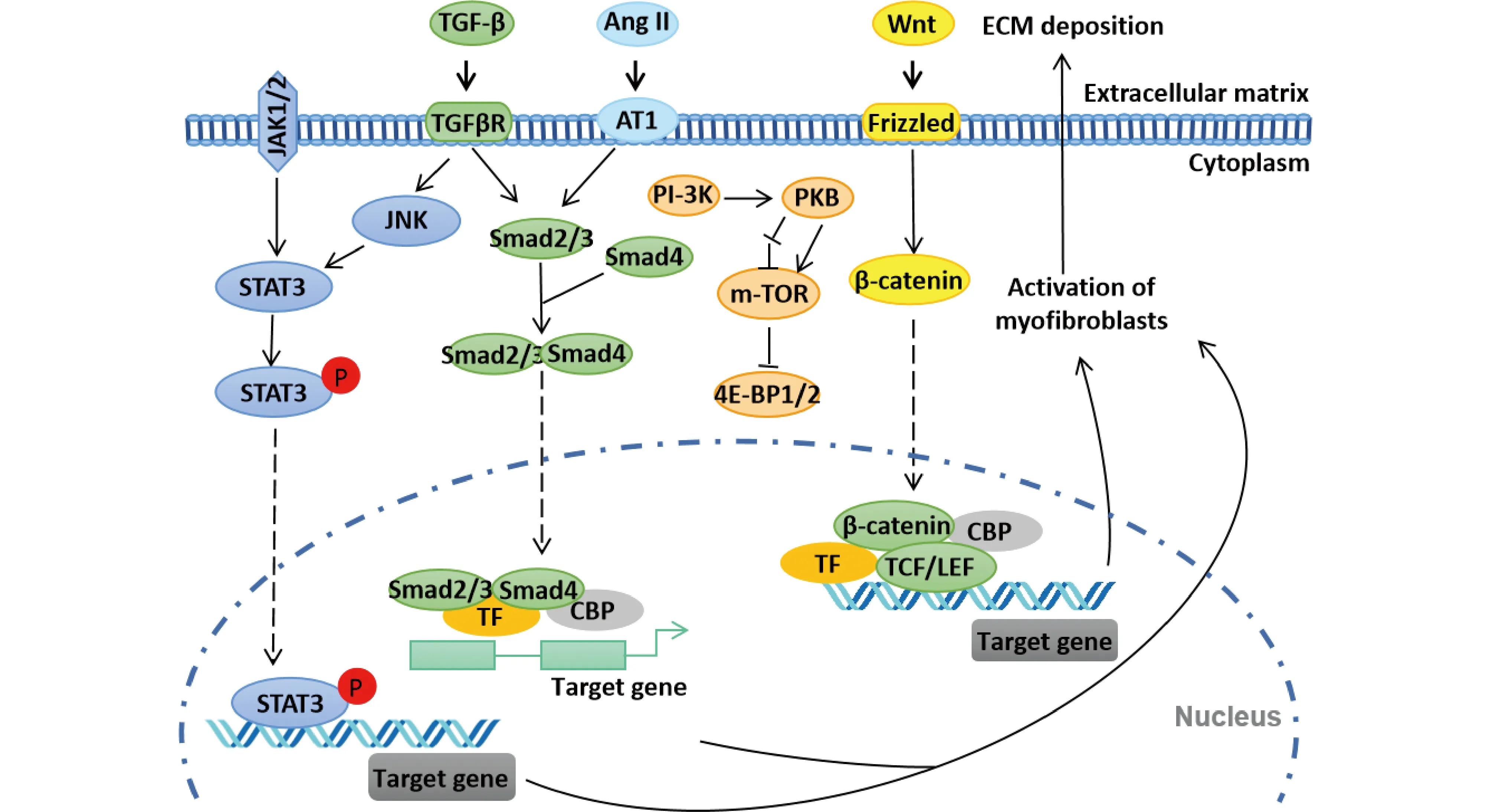
Fig.1 Main signaling pathways of regulatory mechanism during the renal fibrosis Renal fibrosis is a synergic process mediated by multiple signaling molecules involving multiple signaling pathways, including TGF-β/Smad, Wnt/β-catenin, PI-3K/Akt and JNK/STAT3, etc. TGF-β/Smad and Wnt/β-catenin are the two most classical signaling pathways
肾损伤后吸引白细胞、巨噬细胞和肥大细胞等浸润的炎症细胞聚集,导致局部炎症微环境形成。在这种环境中,炎症细胞产生破坏组织的分子,例如活性氧(ROS),并诱导促纤维细胞因子和生长因子的产生,包括转化生长因子-β(transforming growth factor-β,TGF-β)和 Wnt1[24]。TGF-β和 Wnt1与细胞表面相应的受体结合,并发起下游信号转导,最终导致靶基因表达上调,进一步增强MF分化,以及胶原蛋白质和纤维连接蛋白质等ECM的产生和分泌。随着ECM持续沉积,基体的结构发生变化并变硬,形成纤维化。而在分子水平上,肾纤维化是一个由多种信号分子介导的多种信号通路参与的协同过程,包括TGF-β/Smad、Wnt/β-Catenin、PI-3K/Akt、JNK/STAT3和Notch信号通路等。其中,TGF-β/Smad和Wnt/β-catenin是最经典的2个信号通路。
TGF-β/Smad信号是肾纤维化最主要的驱动因素,在各种肾疾病模型的许多细胞类型中都发现,TGF-β/Smad信号通路被激活且TGF-β含量显著上升[17]。TGF-β是引起纤维化的关键细胞因子,主要通过以下几个方面来促进肾纤维化:诱导α-SMA(α-smooth muscle actin,α-平滑肌肌动蛋白)、III型胶原和纤维连接蛋白质等间充质标记物的表达,刺激ECM合成;防止ECM降解;介导肾小管上皮细胞经历上皮细胞-间充质细胞转分化(epithelial-mesenchymal transition,EMT)或肾小管内皮细胞经历内皮细胞-间充质细胞转分化(EndMT),以及直接诱导其他肾细胞[27,28]。
在Wnt信号激活的细胞中,β-联蛋白(β-catenin)在胞浆中不断积累并最终易位至胞核内,成为T细胞因子(T cell factor,TCF)的转录共激活因子,激活促纤维化基因,例如Snail1和MMP7(matrix metalloproteinase 7,基质金属蛋白酶7),使它们大量表达,加速成纤维细胞转化为肌成纤维细胞[29]。Wnt/β-Catenin信号参与调节肾小球硬化和足细胞功能障碍。
4 叉头框转录因子O1与肾纤维化
Luo等[17]研究发现,氧化应激在多种因素诱导的肾纤维化的开始和进展中发挥重要作用,可以促进足细胞凋亡、节段性肾小球硬化和肌成纤维细胞激活,导致肾小管间质ECM重塑。事实上,氧化应激在许多疾病中的作用都归因于ROS的积累。在正常人体中低水平的ROS是一直在产生的,而且无害。但是,许多过量和强烈的刺激物会导致抗氧化系统功能障碍和ROS的积累,ROS会直接损伤蛋白质、氨基酸和核酸等分子,从而导致毒性和氧化应激,最终会导致炎症、纤维化、功能障碍和凋亡[30-32]。
因此,作为已确定的抗氧化因子,FoxO1在肾纤维化的发展过程中必定发挥了重要作用。目前的一些研究也证明了这一点:在糖尿病肾病中,FoxO1/TXNIP-TRX激活的调节作用可以通过抑制细胞ROS的产生来保护高糖诱导的肾近端管细胞损伤[14];给药自由基清除剂TEMPOL,通过调节PI-3K/Akt/FoxO1信号传导来预防肾损伤[33]。
FoxO1可通过多种信号通路调控肾纤维化的发生和发展,这部分将详细介绍FoxO1在肾纤维化中的作用及其调控(Fig.2)。
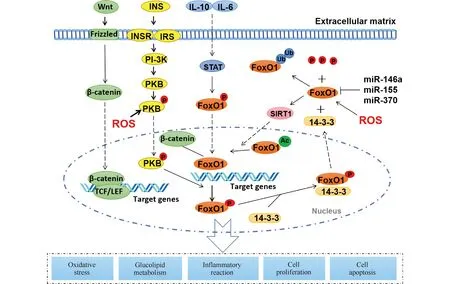
Fig.2 Contribution and regulation of FoxO1 during the renal fibrosis The activity of FoxO1 is primarily regulated by post-translational modifications including phosphorylation, acetylation and ubiquitination. FoxO1 is involved in the pathogenesis of renal fibrosis through multiple signaling pathways, including PI-3K/Akt, SIRT1, STAT, and Wnt/β-catenin signaling pathways
4.1 FoxO1/STAT信号调节TIF和肾小管凋亡
在动物模型中,STAT1的激活与肾小管间质纤维化(TIF)和EMT密切相关[34];而在高糖条件下,FoxO1的再表达降低了系膜细胞STAT1的表达[35]。此外,沉默STAT1还能逆转高糖诱导的足细胞损伤,与位于细胞核中的FoxO1介导的氧化应激有关[36]。Huang等[37]通过小鼠肾特异性过表达Pax2-FoxO1模型发现,肾特异性过表达Pax2-FoxO1使导致糖尿病小鼠肾功能损害的p-STAT1的表达显著降低,从而延缓肾的TIF和凋亡;同时,在HK-2细胞中,FoxO1-KD促进p-STAT1的表达,导致EMT和固有凋亡通路的激活。相反,在高糖条件下,FoxO1-KD明显减弱HK-2细胞的EMT和凋亡。综上所述,FoxO1对肾内TIF和凋亡的保护作用可能靶向STAT1信号,这是糖尿病肾病长期治疗的一个新策略。
4.2 SIRT1/FoxO1信号调控氧化应激反应和脂肪毒性
沉默信息调节因子1 (silent information regulator 1,SIRT1)/FoxO1是机体调控氧化应激的主要信号通路。SIRT1作为FoxO1上游分子,激活后通过抑制FoxO1磷酸化和乙酰化,增强抗氧化系统[38]。SIRT1 /FoxO1信号活化上调机体SOD(superoxide dismutase,超氧化物歧化酶)含量,抑制氧化应激反应,改善缺血再灌注引发的骨骼肌损伤和线粒体功能障碍,并减轻严重烧伤大鼠急性肾损伤以及衰老导致的肾足细胞功能障碍[15,39]。Meng等[40]研究表明,SIRT1可通过去乙酰化激活FoxO1,减轻H2O2所致的细胞氧化应激损伤,抑制成骨细胞凋亡。Yang等[41]研究发现,TMZ通过FoxO1去乙酰化,减轻SIRT1依赖型途径中的氧化应激,从而抑制高糖对EMT的诱导作用。此外,脂质代谢产物异常触发细胞功能障碍,称为脂肪毒性。SIRT1丢失与PPARα(perixisome proliferation-activated receptor alpha,过氧化物酶体增殖物激活受体α)减少引起的脂质代谢异常有关[41]。一些临床和动物研究表明,脂肪毒性会诱导肾疾病的发生和进展,包括糖尿病肾病[42-43];FoxO1亦可提高SIRT1的表达水平[44]。因此,可以通过SIRT1/FoxO1信号调控氧化应激反应和脂肪毒性来调节肾纤维化。
4.3 FoxO1/β-Catenin抑制Wnt/β-Catenin信号通路
在肾纤维化中,骨髓来源的巨噬细胞通过TGF-β诱导的巨噬细胞-肌成纤维细胞转变(MMT)转化为肌成纤维细胞[1]。激活Wnt/β-Catenin 信号通路引起足细胞功能紊乱,进而引起毛细血管壁塌陷以及 ECM沉积,最终导致肾小球硬化[45]。TCF和FoxO1是与β-联蛋白相互竞争性结合的2个关键蛋白质,导致TGF-β对靶细胞的作用不同,甚至相反[20]。当β-联蛋白与TCF结合后会激活许多靶基因的转录,包括结缔组织生长因子(connective tissue growth factor,CTGF)和纤维连接蛋白质等,促进肌成纤维细胞增殖,进而导致ECM不断沉积,加重肾损伤;而当β-联蛋白与FoxO1结合后,会导致细胞周期阻滞,促使细胞在氧化应激状态下存活。小分子抑制剂ICG-001可以阻止β-Catenin/TCF相互作用,将β-联蛋白转移到与FoxO1结合,从而减少β-Catenin/TCF介导的促纤维作用,同时增强FoxO1对调节性T细胞的作用,从而增强TGF-β的抗炎作用[23]。因此,促进β-Catenin/Foxo1结合将抑制β-Catenin/TCF的相互作用,从而阻止TGF-β的促纤维化作用,增加自身的抗炎功能。此外,β-Catenin/Foxo1对TGF-β1诱导的促纤维化蛋白质表达也有抑制作用[23]。
4.4 PI-3K/Akt/FoxO1通路调节炎症反应及糖脂代谢
PI-3K/Akt信号通路主要通过Akt下游蛋白质调控炎症介质的表达(例如mTOR、GSK-3β、eNOS和FoxO1等)来调控炎症反应[46]。PI-3K/Akt/FoxO1信号分子活化后,通过激活CREB(cAMP-response-element-binding protein,cAMP反应元件结合蛋白)、NF-κB p65和AP-1(activator protein-1,激活蛋白-1)等转录因子,进而影响促炎、抗炎反应平衡和免疫应答,最终影响休克、脓毒症和缺血再灌注损伤等炎症疾病的发生和发展[47]。因此,可以通过抑制PI-3K/Akt/FoxO1信号通路的激活,减少炎症因子的产生,从而减缓炎症反应和纤维化的程度。此外,胰岛素是调控机体内部糖脂代谢的重要激素,而PI-3K/Akt信号通路是胰岛素传导的主要通路。FoxO1是其下游转录因子,受胰岛素信号负调控[20]。因此,FoxO1可以通过PI-3K/Akt信号通路来增强自噬因子和糖异生关键酶的表达,从而调节脂肪和葡萄糖的代谢[9],从而在糖尿病肾病中发挥重要作用。最新研究表明,在未对侧肾切除术的单侧缺血再灌注损伤的小鼠模型中,Akt1的缺失导致肾纤维化和肾小管去分化减弱,这个现象独立于 TGF-β1/Smad 信号,反而与糖原合成酶激酶-3β、Snail和β-联蛋白途径有关[48],表明FoxO1可能在这一过程中发挥了作用。
5 问题与展望
综上所述,FoxO1与肾纤维化密切相关,可以通过上述的多种信号通路参与肾纤维化的进程。另外,关于FoxO1与TGF-β/Smad通路在肾纤维化中的研究目前几乎没有。但相关研究表明,在前列腺癌细胞PC3中,过表达FoxO1可以上调Smad4 mRNA和蛋白质的表达水平,且过表达Smad4显著增强FoxO1对PC3细胞增殖和迁移的抑制作用[49];在软骨分化过程中,TGFβ通过 Smad 信号刺激FoxO1核易位,促进FoxO1的表达和活性,并且 FoxO1 是响应 TGFβ 信号传导的 COL2 和 ACAN 表达所必需的[50]。这些研究表明,FoxO1/Smad可能在肾纤维化中发挥了作用。因此,深入研究FoxO1与肾纤维化之间的内在分子机制,有助于为肾纤维化的病理研究提供依据,同时为肾纤维化的预防和治疗提供新的理论支持。
