基于机器学习的蛋白质亚细胞定位预测方法
2022-09-03李佳楠滕小华高兴泉
李佳楠,李 卓,滕小华,高兴泉,唐 友,*
(1.吉林化工学院信息与控制工程学院,吉林吉林 132000;2.吉林农业科技学院电气与信息工程学院,吉林吉林 132101)
蛋白质的亚细胞定位与其功能紧密相关,蛋白质只有处于正确的亚细胞位置才能维持细胞系统的正常运转,蛋白质亚细胞定位研究不仅能够帮助人们了解蛋白质的性质和功能和蛋白质之间的调控机制,还能为人们开发新药物提供有效的参考信息。大多数蛋白质只能在细胞中的1个特定位置(如细胞核、细胞膜)发挥作用,然而一些其他的蛋白质可以在细胞中的几个位置发挥作用。一个蛋白质想要正常的发挥功能,必须处于细胞中的一个或几个特定的位置上,否则该蛋白质就会失效。自后基因组时代以来,产生了大量的蛋白质序列,单纯依靠传统的实验方法进行蛋白质亚细胞定位十分的耗时、耗力。为了更加精准、快速解决蛋白质亚细胞定位问题,人们将研究方法从传统的实验手段逐步扩展到机器学习领域。鉴于此,笔者从蛋白质序列特征的刻画、预测算法、算法评价3个方面阐述现阶段蛋白质亚细胞定位预测的研究进展,总结蛋白质亚细胞定位预测方法方面取得的成果及需要不断完善的3个方面(特征选择、数据处理和改进算法),并提出了未来机器学习在提高预测性能方面的研究重点及重要意义。

图1 蛋白质亚细胞定位预测应用框架Fig.1 Application framework of protein subcellular localization prediction
1 蛋白质序列特征刻画
对国内外相关研究的分析显示,机器学习领域的蛋白质亚细胞定位的发展基本可以分为5个阶段:第1阶段(2006—2010年)的工作主要集中在预测单位点的蛋白质亚细胞位置,但忽略了多位点蛋白质的存在。第2阶段(2011—2013年)的工作主要集中在单位点和多位点蛋白质亚细胞位置的预测,但是大部分为多位点蛋白质开发的技术,在尝试进行预测时却将多位点的问题转化为了单位点的问题。第3阶段(2017—2018年)使用不同的特征提取技术,例如将基因本体(GO)信息融合到通用伪氨基酸组成(PseAAC)中,为多标签蛋白质亚细胞定位开发出了许多的预测器。第4阶段(2018—2020年)是在用预测器对特征提取后的特征向量进行预测之前,使用不同的数据平衡技术处理多标签蛋白质亚细胞定位中的数据不平衡问题。第5阶段(2020—2021年)通过优化机器学习算法以及特征融合来提升预测的准确性,其中具有代表性的算法有深度学习和集成学习。
很多研究者在阐述关于蛋白质亚细胞定位的相关研究时,都用到了Chou的五步法则:①有效构建优质的基准数据集用于模型/分类器的训练与预测;②从蛋白质样本中提取可用于区分不同类别的蛋白质的相关特征;③采用或设计1个优异的分类算法,用于预测各自类别中的不同蛋白质;④选择1个合适的验证方法直观的评价分类模型的有效性;⑤构建1个可公开访问的用户友好型的网络服务器。具体机器学习方法在蛋白质亚细胞定位预测中的应用框架如图1所示。
在进行蛋白质序列特征刻画之前需要构建一个合适的数据集,数据集是算法模型训练和测试的数据基础,它决定了模型训练和测试的效果,因此构建一个合适的数据集十分重要。在构建数据集时应考虑到以下5个因素:①蛋白质序列条数;②需要预测的位点的个数;③是否需要研究多位点定位问题;④特定物种数据集以及基因组数据集的差异;⑤序列同源性大小控制。
目前使用的数据集基本来源于Swiss-Prot数据库和其他的一些关于物种和位置的专门的数据库,如PPDB(plant proteomics database)和NPD(nuclear protein data base)等。
蛋白质是由氨基酸组成的,蛋白质组成形式可由如下公式表示:
=…
(1)
式中,代表蛋白质序列,(=1,2,…,)代表蛋白质序列下的每一个氨基酸。
蛋白质序列原始字母式数据无法直接经过机器学习的方法进行分类和处理,因此需要先将蛋白质的数据转换为一种能够准确地刻画出序列模式信息的离散性数据,再通过机器学习的算法对其进行接下来的分类和处理操作。20种不同的氨基酸残基按照不同的排列组合形成了蛋白质序列,序列中包含了进化特征、序列特征、理化特征等,这些特征对算法的设计和预测结果都会产生影响。提取的特征过少会导致提取后的数据缺失一些重要信息,影响最终预测的结果;提取的特征过多则会导致维数灾难,严重影响算法的效率。因此,如何提取有效的特征并进行融合来提升算法预测的结果仍然是现阶段的核心问题。该研究从序列信息、注释信息和多特征融合3个方面来介绍目前主要使用的特征提取方法:
基于序列信息进行蛋白质序列特征提取的表示方法又可细分为以下3种方法:序列同源性、序列信号、氨基酸组成。
(1)序列同源性。基于序列同源性方法主要通过一些相似性比对工具进行序列间的相似性检验:BLAST(Basic local alignment search tool)、PSI-BLAST是2个很常用的相似性比对搜索工具,PSI-BLAST在BLAST的基础上做了一定的改进,改良过后的PSI-BLAST可对同源性较低的序列之间进行相似性度量。2005年Xie等、2006年Guo等将蛋白质序列同源性信息用于蛋白质亚细胞定位,该方法的缺点为对于一些待测的蛋白质,并不能找到同源性较高的蛋白质序列与之匹配,那么该方法将不再有效。
(2)序列信号。蛋白质的序列上拥有着一部分特殊的子序列,同样特殊的子序列位于蛋白质的N端,而此类子序列被称为分选信号。分选信号的存在会使的蛋白质在功能开展及分选过程当中,转移到特定的亚细胞的位置。目前,已知的分选信号有信号肽、叶绿体运输肽、线粒体转移肽等。序列信号的研究工作一直持续进行,并取得了一定的研究成果,如2000年Emanuelsson等利用N端分选信息预测叶绿体运输肽;2007年Emanuelsson 等开发了基于N端分选信号的蛋白质亚细胞定位方法;2012年Tardif等基于N端分选信号开发了可进行绿藻亚细胞定位预测的工具:PredAlgo。
(3)氨基酸组成。氨基酸是蛋白质序列当中简单直接的特征。ACC的向量表示形式为:
=[,,,…,]
(2)
式中,(=1,2,3,…,)表示蛋白质在中的20中原生氨基酸出现的频率。1994年,Nakashima等最早利用组成蛋白质氨基酸含量的百分率来区分细胞内和细胞外的蛋白质;1995年Chou对ACC的表现形式由原本的20维简化至19维,发现两者是等价的;1998年Reinhardt等在Nakashima和Nishikawa的基础上提出了用氨基酸对进行蛋白质亚细胞定位,构造了蛋白质亚细胞定位第1个人工神经网络。在接下来的几年里,ACC在蛋白质亚细胞定位领域得到了广泛的使用。该方法的缺点为氨基酸组分无法反应序列的局部信息,只能反应序列的整体信息,且氨基酸组分的方法未能考虑到氨基酸的物理化学性质,因此氨基酸组分具有局限性。2000年Chou将序列的顺序因素加入氨基酸组成中进行蛋白质亚细胞定位,发现该方法能有效地提升最终预测结果。
伪氨基酸组成(pseudo amino acid composition,PseAAC)是在2001年由Chou首次提出的一种新的特征提取方法。PseAAC的向量表示形式为:
=[……20+]
(3)
即一组(20+λ)离散因子。PseAAC中的前20个元素域AAC相同,而20+1到20+λ的元素代表不同的序列的顺序相关因子。因子的数量会有所不同,具体取决于所选氨基酸和层级的功能/特性数量。目前,已经开发出4个开放式访问软件:PseAAC、PseAAC-Builder、propy和PseAAC-General。前3个程序作为 PseAAC 的补充,用于计算各种形式的 PseAAC,而PseAAC-General不仅用于生成蛋白质特征向量的所有特殊模式,还用于生成高阶特征向量模式,如功能域模式、基因本体模式和序列进化模式或“PSSM” 模式。现阶段伪氨基酸组成的特征提取方式使用较为广泛。
蛋白质所处的亚细胞位置决定了蛋白质的功能,想要知道蛋白质的亚细胞位置可以从蛋白质的功能信息着手。蛋白质功能域注释信息(functional domain,FunD)。2004、2007年Scott等分别将蛋白质序列上的功能域注释信息用于蛋白质亚细胞定位。功能域注释信息虽然具备较高的可靠性,但同时该方法也有一定的缺陷,即使用功能域注释信息时需要保证功能域数据库中的功能域条目达到一定的量才能确定序列中特定的功能域。
基因本体(gene ontology,GO)是基于GO数据库的一种特征提取方法,包括了分子功能、生物学过程和细胞组件3种基本信息。2010年Qu等、2013年Pacharawongsakda等通过使用GO特征提取,实现了蛋白质亚细胞定位预测精度的显著提高。2018年研究人员通过提取GO特征信息,开发出了一系列用于多位点蛋白质亚细胞定位预测的web服务。尽管GO很重要,但它有以下主要缺点:①提取蛋白质的GO注释信息会产生大量特征,需要进一步处理和过滤才能提取出有区别的特征;②新蛋白质的GO信息不可用,许多研究使用基于同源性的方法来提取这些蛋白质的 GO信息,从而导致提取的信息不准确。
单纯依靠单一的特征提取模型来提取特征是远远不够的,将几个模型结合起来可能会得到显著的性能,但如果里面包含不合适的模型,结果会适得其反,这是由于过拟合影响模型的计算造成的。因此,需要对于不同的数据集要有针对性选择不同的特征提取方法进行融合。
Qu等融合了5种基于氨基酸物理化学性质的特征提取算法,使用过程当中发现分类器性能相比于单个特征提取算法的分类器更为明显。Javed等将29种氨基酸物理化学性质用于伪氨基酸组成方法,通过和SAAC方法的融合,构建出的分类器性能得到显著提升。国内一些学者在特征融合的道路上也取得了一定的成就:2019年,刘清华等基于特征融合思想在Gram-negative和Gram-positive数据集准确率分别达到了89.6%和97.8%。2020年王艺皓等将改进型伪氨基酸组成法、伪位置特异性得分矩阵法和三联体编码法共3种特征提取方法进行融合,在Viral proteins数据集和Plant proteins数据集上分别取得了98.24%和97.63%高准确率。
多特征融合的目的就是为了更好地提高分类器的效率和算法预测的准确度,但同样多特征融合也是蛋白质序列特征提取的重点和难点部分,对于不同的数据集不能采用同种方式进行多特征融合,需要根据数据类型有所区分,选择适合该数据的特征进行融合。目前多特征融合提取蛋白质序列特征的方法是现阶段主要的研究方法之一。
2 预测算法
通常,模型开发面临的一个问题是用于实验的数据集不平衡。细胞内有的蛋白质峰度较高,有的蛋白质峰度较低,这样容易导致样本集中的样本严重不均衡。用于预测的数据集中属于某一个类别的蛋白质数量通常情况下不同于属于其他类别的蛋白质数量,如果不同类别下的蛋白质数量之间的差异很大,这种情况下就会使得分类模型过度分类,即由于属于某个类别的蛋白质样本数量较多,分类器在预测数量较多的类别下的蛋白质时达到的精度可能会更高。
为了解决数据不平衡问题,增强识别交互对的能力,很多的研究者提出了很多不同的方法:1992年Zhang等提出了蒙特卡罗样本扩展方法(Monte Calo sampling approach);1995年Zhang等提出了种子传播方法(Seed-propagation approach);2006年Cai开发出了LogiBoost分类器;2001年Laurikkala提出了NCR(neighborhood cleaning technique)方法用于去除冗余的样本;与NCR类似的还有KNCC方法。以上这些方法一部分是在少数类中添加重复的实例或者从多数的类中裁剪同源样本来使数据集达到平衡,另一部分则为训练样本分配权重来处理数据不平衡问题。研究人员证明了这种线性重采样对训练模型的性能没有多大的提升,因为如果在少数类中添加类似的样本,分类器会识别出这些相似的区域,这种情况下会导致模型训练的过拟合。2002年Chawla等提出了SMOTE算法,SMOTE算法也在数量较少的类中增加额外的样本,但这些样本并不是类似的样本,而是通过沿着属于特定少数类的线的“K个最近邻”来综合收集的。
结果表明,在对不同的数据集进行平衡过程中,SMOTE算法很明显几乎优于其他所有的重采样方法。在后续的很多研究中也证明了平衡后的数据在通过分类器进行分类时的效果要优于未平衡前的数据。
预测算法的优劣对蛋白质亚细胞定位预测的准确性有较大影响。开发一个性能优异的预测器需要达到2方面的条件:其一是进行特征提取时要根据数据的特性合理的提取特征,其二就是需要一个高通量、高准确率的预测算法。在过去的研究期间内,曾出现非常多的预测算法。刚开始的算法设计较为单一,进行分类的精度不够,后逐步开始研究集成机器学习;起初未发现多位点蛋白质时研究重点在单位点的蛋白质亚细胞定位,后来多位点蛋白质的发现研究重点转移至多位点多标记学习问题。虽然现阶段算法预测的准确率不断提高,但仍需要进一步深入的研究。集成学习的方法仍旧是目前及未来研究的重点。接下来介绍几个具有代表性的算法及多标记学习算法:
最近邻算法(nearest neighbor,NN)通过某种距离度量方法判断2个样本之间的距离关系:若距离越近,出现在同一个细胞器中的可能性越大;反之则可能性越小。1996年Horton等提出了KNN(K-nearest neighbor,KNN)分类算法;2004年Huang等使用了模糊KNN方法预测蛋白质亚细胞定位;2006年Chou等通过融合优化的证据理论 K 最近邻分类器(OET-KNN)预测真核蛋白质亚细胞位置;2017年薛卫等基于相似性比对改进KNN的Adaboost集成分类预测算法,在数据集CH317和Gram1253上的最高预测准确率达到了92.4%和93.1%。目前一种解决多标记问题的多标签K近邻算法(ML-KNN)正被广泛使用。
KNN算法的思想是某个样本类别由其附近的个相似样本中的大多数决定。模糊KNN算法在KNN算法的基础上为增加了样本隶属度,这样可以减少KNN算法在运算时的错误率从而提高分类的准确率。
人工神经网络是一种可以进行信息处理的数学模型,在使用过程当中,类似于人类大脑中的神经突触连接结构,模拟人类大脑进行信息的传递以及信息的处理。人工神经网络算法有3个非常显著的优点:第一,本身具备非常强的自我学习功能;第二,具备先进的联想存储功能;第三具备寻找优化解的能力。1998年,Reinhardt等第1次将神经网络用于蛋白质亚细胞定位预测研究;2000年Emanuelsson等采用了人工神经网络的方法进行预测;后来Sun等将概率神经网络用于蛋白质亚细胞定位预测;目前的一些研究中,使用ML-RBF(RBF neural networks for multi-label learning)对多位点的蛋白质进行亚细胞定位预测,并取得了较好的效果。
1995年Vapnik最先提出支持向量机(Support Vector Machine,SVM)的概念。支持向量机方法在使用过程当中能够针对高维模式识别,非线性以及小样本问题进行有效的解决并且具备该方法具备独特的应用优势,能够将其应用到函数拟合等其他的机器学习问题当中。2011年Hua等第1次将SVM算法用于蛋白质亚细胞定位预测;后来一些学者使用SVM算法进行预测并取得了较好的效果;2017年赵南等运用词袋模型结合传统的蛋白质特征提取方法,在SVM分类器上进行分类,在一定程度上提升了预测的准确率;2019年研究人员基于特征融合的思想利用LDA方法进行降维,再利用SVM算法进行分类,在Gram-negative和Gram-positive数据集上取得了较好的预测效果;2020年胡雪娇等提出了一种基于PSO_BFA优化的词袋模型,获得蛋白质序列的词袋特征后放入SVM分类器中有效地提高了蛋白质亚细胞定位预测精度。
集成机器学习即使用多个不同的学习器的方式来解决同一个问题,通过集成学习的方式可以大大提高学习系统的泛化能力。但同样集成机器学习也有其局限性,研究小组将集成学习的方法用于蛋白质亚细胞定位预测:Laurila等提出了一种集成方法(PROlocalizer),它结合了多个专门的二进制定位预测算法;Park等开发了一种线性判别分析 (LDA) 方法 (ConLoc) 来为加权投票分配 LDA 最佳权重;Assfalg等提出了2种集成定位算法:一种是基于预测器的预测精度等级的评分投票方案,另一种选择J48决策树(DT)分类器作为集成方案;Shen等提出了1种两层决策树方法进行蛋白质亚细胞定位预测。这些方法大都数集成了10种或者更多的方法用于预测,但没有考虑它们之间冗余或互补的关系,导致集成算法的计算量很大,后来Lin等提出的极简集成算法有效地解决了这个问题,2017年薛卫等基于相似性比对改进KNN的Adaboost集成分类预测算法在数据集CH317和Gram1253上的最高预测准确率达到了92.4%和93.1%。
现阶段研究的重点就在于多位点蛋白质亚细胞定位预测问题,多位点蛋白质亚细胞定位换种方式讲就是一种多标记学习的问题,对于多标记学习问题可以按照算法分为2类,即问题转换型和算法适应型。问题转换型算法即通过二分类算法解决分类问题的方式解决多标记学习问题;算法适应型算法直接构造一种有效的算法解决多标记学习的问题。
问题转换型。BR (binary relevance ) 方法是一种十分经典的问题转换型方法,它的特点是简单有效且算法复杂度低,在多位点蛋白质亚细胞定位中多以SVM作为基本算法。BR方法的缺点:BR方法虽可以并行运算,但却没有考虑标记之间的相关性。
CC(classifier chain) 方法在使用过程当中,注重标记样本之间存在的关联性,并在此基础之上进行模型构建,但它不能进行并行运算且标记的训练顺序具有随机性,对预测器性能影响较大。
ECC(Ensemble Classifier Chains)方法通过集成学习的方法解决了标记顺序带来的随机性问题,但同样也带来了计算量大等问题。
算法适应型。ML-KNN(Multi-label k-nearest neighbor) 方法是一种基于实例的多标签分类方法,由传统的KNN算法发展而来。ML-KNN在训练集中识别K近邻并捕获每个实例的相关统计信息,采用最大后验概率准则来预测未知的标签。试验结果表明,ML-KNN算法有较好的效果。
ML-GKR(Multi-label gaussian kernel regression)在很多现有的预测器中都有使用,且达到了不错的效果。
ML-RBF(RBF neural networks for multi-label learning)方法与传统的径向基函数存在直接关系,算法中第一层主要针对标记进行聚类,并对这些标记进行分析,分析后聚类中心作为算法的基函数原型向量,通过最小化残差平方和计算,得到神经网络第二层。
3 算法评价
算法预测的准确率是衡量算法优劣的重要指标,而评价一个分类算法性能好坏的重要指标是对该算法的性能评估。目前在测试过程当中主要应用的验证方法有3种,分别为独立数据集测试、二次抽样测试以及刀切法测试。其中,刀切法(Jackknife test)是目前最为普遍采用、最被人们认可的验证测试方法。
Jackknife测试方法是Maurice Quenouille在1949年提出的一种再抽样方法,jackknife方法主要将数据集的每个样本依次挑选出来,挑选出来的样本用作测试,其余的样本用作训练,这样做的好处是每一个样本都有可能被作为测试,这样能最大程度上减小误差,极大增强了测试的客观性,是现阶段使用的最多且最为客观的验证测试方法。在独立数据集测试中,将训练集和测试集作为2个互相独立的蛋白质数据库;而二次抽样测试(sub-sampling test)则是从训练集中轮流抽取一个样本作为测试集,将其余的样本作为训练集。
蛋白质亚细胞定位分类器的性能通常可以从以下几个指标进行评价:
总体准确率ACC:
总体准确率ACC:

(5)
敏感度SN:

(6)
特异性SP:

(7)
精度PV:
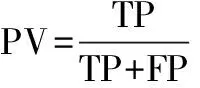
(8)
马氏相关系数MCC:

(9)
式中,TP为分类模型正确预测的正样本数量;FP为分类模型错误预测为正类的负样本数;TN为分类模型正确预测为负类的负样本数;FN为分类模型错误预测为负类的正样本数;MCC取值范围为0~1,MCC的取值越高说明算法性能越好。
当一个算法通过验证具有优异的性能及预测结果后,应当考虑建立一个web服务供其他的研究人员使用。目前已有大量的蛋白质亚细胞定位预测服务器提供了相应的web服务,其中包括单位点和多位点预测服务器,表1列出了一些比较有影响力的预测服务器:

表1 部分有影响力的预测服务器列表Table 1 List of some influential prediction servers
4 结语
蛋白质亚细胞定位预测研究是生物信息学领域研究的重点问题之一,面对海量的蛋白质序列数据,研究出如何利用计算机技术实现高效、精准的蛋白质亚细胞定位预测是十分必要的。经过几十年的发展,亚细胞定位预测的方法不断地完善,主要体现在以下3个方面:
(1) 刻画蛋白质序列的信息越来越丰富。面对越来越复杂的蛋白质序列数据,单一特征提取的方法已不能有效地将这些蛋白质区分开来,多特征融合的方法是现阶段改进识别效果最有效的手段。
(2) 数据集平衡后提升预测精度。数据集中属于某一个类别的蛋白质数量通常情况下不同于属于其他类别的蛋白质数量,如果不同类别下的蛋白质数量之间的差异很大,就会使得分类模型的过度分类。后续研究者研究发现,数据集平衡后能有效提升预测精度。
(3) 识别算法越来越复杂。从初期的简单分支算法到KNN、神经网络、支持向量机和深度学习的使用再到现阶段很多预测器使用的集成算法,这些复杂的算法有效克服了数据复杂度增加带来的困难,大大提升了预测精度。
通过对以上3个方面有关完善蛋白质亚细胞定位预测方法的总结,提出了4点关于未来机器学习在蛋白质亚细胞定位预测方面的重要研究方向及研究意义:
(1) 特征融合时若包含不合适的模型往往会影响模型的计算,因此如何选择更加合适的模型进行融合依旧是研究热点。多个合适的模型进行融合后会覆盖数据的大部分重要信息,对预测结果有十分积极的影响,是蛋白质亚细胞定位预测研究的重点部分。
(2) 多位点蛋白质数据往往会存在很严重的数据不平衡现象,因此如何有效解决数据不平衡问题来提升预测精度成为研究重点之一。解决数据不平衡问题,增强识别交互对的能力,有利于提升整体预测效果,达到提升预测精度的目的。
(3) 蛋白质之间存在一定的相关性,如何利用好蛋白质之间的相关性提升预测精度是重要的研究方向。有效利用蛋白质之间的标记相关性,能更好提升蛋白质亚细胞定位的预测性能。
(4) 一个性能优异的预测算法能充分的反应分类问题的本质,能很好地提升预测的精度。集成学习方法是现阶段研究的重点内容,因此开发高效的集成学习方法也是重要的研究方向。
