基于LASSO回归和多层感知的癌组织RNA-Seq数据分类算法研究
2022-08-31颜滢李文敬李松钊
颜滢 李文敬 李松钊


摘要:目的:为了解决癌症基因RNA-Seq(RNA-Sequencing,转录组测序技术)技术每次测序过程产生海量高分辨率、高维、高冗余的数据,给基因表达数据分类带来困难的问题。方法:提出了一种基于LASSO(Least Absolute Shrinkage and Selection Operator,LASSO)回归和多层感知的癌组织RNA-Seq数据分类算法。首先,从TCGA数据库获取十个疾病的基因数据集并对原始RNA-Seq的基因表达谱基因数据进行数据清洗和标准化处理,去除重复的基因,选取表达量最大的基因并将数据做标准化处理。其次,采用LASSO回归的方法对处理后的数据进行降维和特征提取,获得与疾病标签最相关的特征基因集。最后,运用多层感知器神经网络(Multilayer Perceptron,MLP)模型对特征基因进行学习和训练,实现有效地识别和分类。实验结果:实验表明,该算法在10种癌细胞基因测试数据集中分类总准确率达到99.8%,高于LASSO-CNN分类模型的总准确率98.9%和LASSO-BP神经网络分类模型的总准确率99.4%。结论:该算法克服了转录组测序数据量大、特征多、数据差异大的缺陷,是一种有效的癌症基因表达测序分类新算法。
关键词:RNA-Seq;LASSO回归;特征提取;多层感知器神经网络;基因表达;TCGA数据库
中图分类号:TP3 文献标识码:A
文章编号:1009-3044(2022)19-0091-03
转录组测序技术(RNA Sequencing,转录组测序技术)能够对生物的转录本进行检测,确定哪些变异在癌症样本中有表达,通过分析基因表达差异识别出变异基因或癌症基因,在肿瘤疾病的诊断和治疗起着重要作用,具有重要的科学意义与应用价值。但是,转录组测序技术可以在一次实验中获取大规模的基因表达谱数据[1],若要在海量的信息中识别疾病相关基因,使RNA-Seq技术在肿瘤疾病的诊断和治疗中发挥重要作用,则要引用特征选取和机器学习的方法。为研究高效率、高准确率的基因分类算法,本文提出一种基于LASSO回归和多层感知的癌组织RNA-Seq数据分类算法,在一次对癌症样本RNA-Seq测序后,可直接将结果进行识别、预测、分类。
为了解决高维基因数据的特征筛选和分类问题,1996年Robert Tibshirani[2]提出的LASSO回归算法为基因特征数据的提取提供了技术支持,并逐渐应用到生物信息学领域。對于基因数据的特征筛选和分类问题,张靖等人[3]提出一种基于迭代Lasso的信息基因选择方法,采用改进的Lasso方法进行冗余基因的剔除以获得基因数量少且分类能力较强的信息基因子集,并使用支持向量机(SVM)、K近邻(KNN)、决策树C4. 5和随机森林Random Forest4种分类器进行分类。张靖、张玉红等人[4]提出K-split Lasso特征选择方法,其基本思想是将数据集平均划分为K份,分别使用Lasso方法对每份进行特征选择,而后将选择出来的每份特征子集合并,重新进行特征选择,得到最终的特征基因,最后采用支持向量机进行分类。Ma[5]等人结合K-means和Lasso方法对基因表达谱数据进行特征选择和预测模型构建,取得了较好的效果。
1本文算法原理
1.1 LASSO回归原理
在样本基因数据中引入的特征太多,主成分分析法选择将一些原始数据丢失[6],而这些数据可能含有对样本差异的重要信息,这就会对区分样本类别的结果产生影响。采用LASSO回归(Least Absolute Shrinkage And Selection Operator)更适用于处理一次RNA-Seq技术测序所产生的数据,LASSO回归通过参数缩减拟合广义线性模型的同时进行变量筛选,从而达到降维和选取特征基因的目的[7]。这个方法能够保留原有的基因特征属性,选取关键特征,可直接用于特征建模分析。
以提取多种癌症组织样本特征为例:给定[n]个疾病样本[{(X1,Y1),…,(Xn,Yn)}],自变量[X=(x1,x2,…,xn)T∈Rm*n]为基因数据矩阵,[xn∈Rm]为m维数据样本,包含m个特征,响应变量[Y=(y1,y2,…,yn)T∈Rn],[Y]为疾病标签,自变量[X]对响应变量[Y]进行线性回归,约束[λ=(λ1,λ2,…λt)]不超过阈值[e]。
设本实验目标函数为:
LASSO回归优化目标是令代价函数(cost function,或称为损失函数,lost function)最小,
[min L(λ)=12nj=1n(yj-λTxj)2+μj=1t|λj|subjecttoj=1t|λj|≤e](1)
n为样本个数,[μ]为正则化参数,[t]为参数个数。随着[μ]的增大,各变量的系数逐渐趋于零。
1.2 多层感知器
多层感知器(Muti-Layer Perception,MLP)是一种前馈式人工神经网络,是目前最成熟的人工神经网络之一。它由三层结构组成,分别是:输入层、隐藏层和输出层。MLP神经网络能学习和存储大量输入-输出模式的映射关系,被广泛应用于图像,自然语言处理,生物信息领域识别、预测、分类[8]。
2 多层感知的癌组织RNA-Seq数据分类算法的构建
2.1 获取数据集与基于R语言的数据处理
2.1.1 数据集的获取与数据预处理
本次实验样本基因数据来源于TCGA数据库,TCGA是关于癌症方面的最大的公共数据集[9],为研究肿瘤学的人们提供了便捷的数据获取平台。本实验使用3782个样本进行建模,每个疾病样本包含25190个基因,原始数据无法直接用于模型训练,因此要进一步对数据进行处理。
从数据库获取到的基因数据集为COUNT矩阵,将COUNT矩阵导入R,把基因ID转换为Gene symbol,去除重复的基因,选取表达量最大的基因,这些基因将用于做数据标准化。
2.1.2 数据编码:One-Hot
本实验序列的标签将采用One-Hot的方法进行编码。用LIHC、STAD、BRCA、DLBC、ESCA、GBM、OV、PAAD、LUAD、UCEC這10种癌症基因数据进行分类,并将患病样本所对应的疾病作标签。
2.1.3 数据标准化
数据标准化的目的主要是消除测序数据的技术偏差[10],各个样本基因数据间的测序深度和基因长度处于相同的水平,从而使我们得到具有生物学意义的基因表达量变化。本实验则采用了文献[11]的方法,使用基于R语言的voom函数对RNA-Seq基因数据标准化处理。
2.2 基于LASSO回归的降维及特征提取的实现
LASSO回归的核心思想是将不相关的特征系数变为零,从而筛选出含有特征基因变量。具体实现如下:
(1)构造一个从200的-5次方到200的2次方的等比数列,这个等比数列的长度是200个元素,[λ]即这200个元素中不同的值。
(2)给定一个变量alphas,用于进行交叉验证的正则化参数。令alpha=[λ],采用十折交叉验证的方法找出最佳的alpha值,迭代1000次。
(3)调用最佳正则化参数下建立的模型系数,输出相关系数不为零的特征。
(4)记录相关系数不为零的特征,用于构造新的数据集。
(5)划分数据集,设定一个随机种子,在任意带有随机性的类或函数里作为参数来控制随机模式,得到新的数据集按7:3的比例划分,得到比例为7:3的训练集与测试集。
本实验从25190个基因中提取到与标签最相关的1414个特征基因及其表达量这些数据将用于模型训练。
2.3 模型训练
参数设置:实验中MLP神经网络的激活函数设置为relu函数,隐藏层设为3层,每一层隐藏层的神经元设置为500,第一层隐藏层的学习率设置为0.1,第二、第三层的隐藏层学习率设置为0.2。
实验环境:Intel CPU 3.20 GHz处理器,8 GB内存的PC机,Windows 10操作系统,PyCharm 2020.3.3开发环境。
①信息前向传播
设[ol]=[(ol1,ol2,....,oln)T]为第[l]层的输出,[l]=(1,2,3,4,5),n=(1,2,...,500)
当[l]=1时,
[oli]=[xi] (2)
当[l]≥2时,
[ol=Wl*ol-1+bl] (3)
当[l]=5时,此时为输出层:使用多分类函数softmax计算得到输出层的输出:
[y=exp(o4)n=1500exp(o4n)] [(4)]
隐藏层间的激活函数relu:
relu[(x)=max(0,x)] (5)
②信息反向传播
设代价函数(cost function)为[E],N为训练样本个数:
[Etotal=12Ni=11||yi-xi||2] [(6)]
优化目标为确定W(权值)和b(偏置)使得损失函数[E]最小,采用梯度下降法更新参数的公式为:
[Wl=Wl-δNi=1N?EiWl] [(7)]
[bl=bl-δNi=1N?Eibl] [(8)]
式中:[δ]为学习速率,取值范围(0,1]。
3 实验结果与分析
本文采用BP网络、CNN网络做对比实验用于验证本文算法的优势。
3.1 实验结果
3.2 实验结果分析
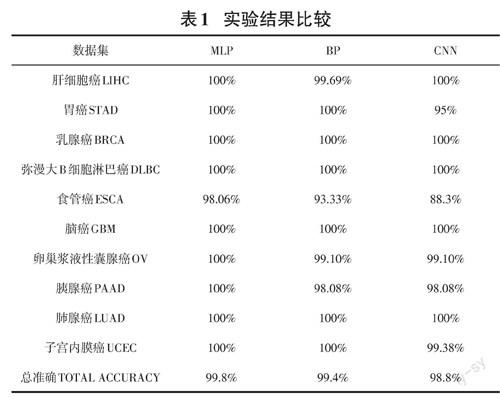
分别构建基于MLP、BP和CNN的分类模型,得到RNA-Seq基因样本在3种模型下的识别准确率如表1所示。根据表1得知,在MLP模型中,准确率为99.8%,分类效果较为理想;在BP模型中,准确率为99.4%;在CNN模型中,准确率为98.8%,分类效果相对较差。根据上述的分类结果可知,MLP模型能够使用多类别基因数据识别方式对RNA-Seq数据样本进行有效区分,且效果最佳。
4 结束语
本文提出了一种基于LASSO回归和MLP模型构建对多种癌组织样本RNA-Seq基因序列的分类算法,本算法增加了训练样本数量,与其他神经网络的分类方法相比具有较好的分类效果,且优于文献[9]的分类算法准确99.3%。在LASSO回歸算法的基础下,提取出样本特征,为多层感知器提供了输入数据,增加了模型分类的准确率和进一步提高了泛化能力。基于LASSO回归的多层感知器模型的识别的准确率为99.8%,符合多种癌症RNA-Seq基因序列的分类需求,同时也为其他基因数据分类方法提供借鉴。
参考文献:
[1] DERISI JL, IYER VR, BROWN PO. Exploring the metabolic and genetic control of gene expression on a genomic scale[J]. Science, 1997, 278(5338): 680-686.
[2] Tibshirani R. Regression shrinkage and selection via the lasso [J]. J Royal StatSocSer B Methodol, 1996, 58(1): 267-288.
[3] 张靖, 胡学钢, 李培培, 等. 基于迭代Lasso的肿瘤分类信息基因选择方法研究 [J]. 模式识别与人工智能, 2014,27(1): 49-59.
[4] 张靖, 胡学钢, 张玉红, 等. K-split Lasso: 有效的肿瘤特征基因选择方法 [J]. 计算机科学与探索, 2012, 6(12): 1136-1143.
[5] MA SG, SONG X, HUANG J. Supervised group Lasso with applications to microarray data analysis [J].BMC Bioinform, 2007, 8: 60.
[6] 纪荣芳. 主成分分析法中数据处理方法的改进[J].山东科技大学学报(自然科学版), 2007,26(5): 95-98.
[7] 王福友,白冰,徐平峰.基于SIS的基因表达数据分析[J].长春工业大学学报, 2017, 38(5): 417-420.
[8] 张驰,郭媛,黎明.人工神经网络模型发展及应用综述[J].计算机工程与应用,2021,57(11):57-69.
[9] 蒋文妍.基于RNA-Seq数据的癌症标志物研究[D].天津:天津工业大学,2020.
[10] Conesa A,Madrigal P,Tarazona S,et al.Erratumto:a survey of best practices for RNA-Seq data analysis[J].Genome Biology,2016,17(1):181.
[11] YANG YH, DUDOIT S, LUU P, et al. Normalization for cDNAmicroarray data: a robust composite method addressing single and multiple slide systematic variation [J].Nucleic Acids Res,2002, 30(4): 15.
收稿日期:2022-03-20
基金项目:国家自然科学基金(61866006)
作者简介:颜滢(1997—),女,广西灵山人,硕士,主要研究方向为生物信息计算、智能计算;李文敬(1964—),男,广西南宁人,教授,主要研究方向为并行计算、智能计算;李松钊(1994—),男,广西灵山人,硕士,主要研究方向为智能计算。
