PROKR2基因突变致卡尔曼综合征1例及文献回顾
2022-08-29张俊平徐积兄
张俊平,徐积兄
(南昌大学第一附属医院内分泌代谢科,南昌 330006)
卡尔曼综合征(KS)是以特发性低促性腺激素性性腺功能减退症(IHH)和嗅觉减退为特征的内分泌疾病。患者性腺发育不全和嗅觉功能障碍的严重程度因致病基因的不同而有所差异。PROKR2基因是KS经典的致病基因,本文报告1例由PROKR2单基因突变导致的KS,并回顾总结文献中该类患者的病例特点。
1 临床资料
1.1 病史及体格检查
患者男性,17岁,因“自觉阴茎短小5年”就诊,患者于青春期期间,自感发育迟缓、第二性征不明显。既往史:无出生窒息史,身体良好。家族史:父母非近亲婚配,双方家族均无类似病史。体格检查:身高170.5 cm,体重60 kg,BMI 20.64 kg·m-2,身材比例匀称,神志清楚,无明显运动及智力发育落后,无特殊面容。专科检查:Tanner分期双乳B2期、睾丸G1期,阴茎长约2 cm,上翻包皮可外露龟头,双侧睾丸位于阴囊内。嗅觉正常,听力正常,双眼高度近视(皆600度),无视觉障碍,无色盲,无喉结,无体毛生长,无唇腭裂,无牙龈缺损,无痤疮,无手部镜像运动。
1.2 辅助检查
1)一般检查:血常规、肝肾功能、电解质、同型半胱氨酸、肿瘤标记物无异常。2)激素检查:雌二醇<11.8 pg·mL-1,睾酮21.23 ng·dL-1,泌乳素4.15 ng·mL-1,孕激素0.32 ng·mL-1,17-α羟化酶0.59 ng·mL-1,雄烯二酮0.64 ng·mL-1,脱氢表雄酮112 μg·dL-1,双氢睾酮83.404 pg·mL-1,骨钙素65.79 ng·mL-1,人绒毛膜促性腺激素、胰岛素生长因子、甲状腺功能三项、甲状旁腺激素、ACTH、皮质醇节律均正常。3)GnRH激发实验:注射戈那瑞林25 μg后FSH及LH变化见表1。4)影像学检查:阴囊彩超示双侧睾丸微石症,双侧睾丸回声不均匀,大小左侧25 mm×16 mm×12 mm,右侧24 mm×15 mm×11 mm。乳腺彩超示双侧乳房可见腺体组织。肝胆胰脾彩超、双肾及肾上腺彩超未见明显异常。手腕DR检查示骨龄14岁。垂体磁共振示未见明显异常。5)染色体核型:46,XY,320~400常规G显带未见明显异常。6)基因检查:提取患者及其母亲的血液样本,外送至北京迈基诺基因科技股份有限公司,行全外显子基因检测V4分析,患者本人采用高通量基因组测序(NGS),母亲行Sanger验证。患者本人为位于PROKR2基因(NM_144773;exon3)的杂合性突变c.533G>C(p.W178S),由于患者母亲无该变异基因,且患者父亲未采样,故变异来源暂不明确(表2)。
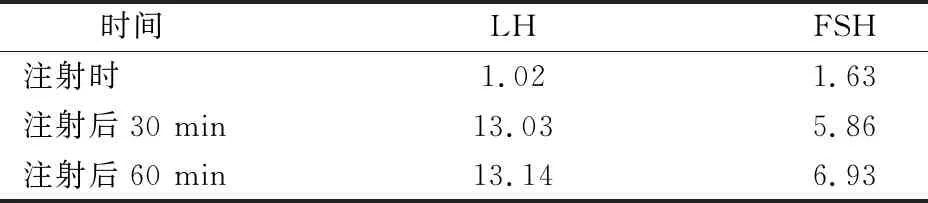
表1 注射戈那瑞林25 μg后FSH及LH变化 mU·mL-1

表2 患者基因分析结果
1.3 诊断及治疗
患者小阴茎,性腺组织与外生殖器不相配,第二性征缺乏,结合基因检测结果,诊断为KS。给予睾酮替代疗法,嘱患者口服十一酸睾酮软胶囊(第1周40 mg qd,之后40 mg bid)治疗。口服3个月后,门诊复查FSH 2.44 mU·mL-1,LH 3.43 mU·mL-1,睾酮T(TSTO)17.70 ng·dL-1,遂嘱患者调整用药为40 mg tid,并定期门诊随访。
2 讨论
KS是一种罕见的内分泌疾病,其发病率在男性约为1/1万,女性中约为1/5万[1]。KS是IHH的一种特殊类型,其临床表现是在缺少促性腺激素释放激素(GnRH)的同时常常伴有嗅觉功能的障碍。近年来随着对KS的深入了解,现普遍认为其致病机制为:嗅神经元和GnRH神经元在胚胎发育期间,均来源于嗅板,两者在胚胎发育过程中会一起迁移至大脑,而由于相关的基因突变扰乱迁移过程,致使两者未到达大脑特定区域发挥功能,从而产生性发育不全及嗅觉障碍共存的临床表现。截至目前,已报道过的相关基因突变已达20多种,不同的基因突变可通过不同的方式遗传,部分常见的KS相关基因及其遗传方式如下:X连锁隐性遗传(可由ANOS1和DAX1基因引起)、常染色体显性遗传(可由CHD7、FGFR1和SOX10基因引起)和常染色体隐性遗传(可由GNRH1和KISS1基因引起)[2]。
本例患者于NGS检测中发现的PROKR2基因,在2006年被DODÉ等[3]首次通过使用直接候选基因的方法,确定了其为KS的疾病相关基因。截止目前,PROKR2基因已有30多种突变位点被相继报道(A51T、R85C、D112Y、S202G、V331M、M64V、V317L、R357W、S188L、V297I、V158I、R248Q、R85L、Q210R、M323I、T260M、V274D、M111R、Y113H、R80C、R85G、R85H、R164Q、G234D、W251L、R270C、N325K、V115M、R117W、W178S、P290S、L173R、R268C、V334M)[4]。由PROKR2单基因突变导致的KS具有部分与其他基因所致KS不同的特点,总结如下。1)KS的1个主要临床特征可表现不全,患者大致分为:①患有IHH,但嗅觉正常;②嗅觉缺失/低嗅觉,但青春期发育和性腺功能正常;③没有症状,嗅觉正常,青春期发育完全;④嗅觉缺失和IHH皆有。2)其他非生殖系统表现的患病率相对较低,如先天性上睑下垂/眼神经传导障碍、肾脏发育不全/马蹄形肾、输精管发育不全等。3)患者肥胖和睡眠障碍的患病率可能增加,这是由于PROKR2可在控制食物摄入和调节昼夜节律中发挥作用[5]。本例为男性患者,临床表现主要为IHH,嗅觉及其他系统表现均无特殊,是较为典型的单PROKR2基因突变的KS病例。
男性KS患者的治疗,主要有3种药物[6-7],分别为性类固醇激素、促性腺激素和脉冲性GnRH给药。治疗方案的选择主要取决于治疗目标(即诱导和维持第二性征/诱导和维持生育能力)。本例患者为青少年,暂不立即需要生育能力,使用睾酮替代疗法是最实际的选择。由于患者青春期发育不全,因此初始治疗应当从低剂量开始,后逐渐增加至成人剂量,以确保患者第二性征发育、具备正常的性功能、维持适当的肌肉和骨骼力量。当患者有生育需求时,可使用促性腺激素或脉冲性GnRH疗法来实现男性的生育潜力。
KS的早期诊断尤为重要,然而患者的临床特征通常在青春期后才表现出来,且患者普遍年龄偏小,对自己的症状往往难以启齿,不肯告知家长,加之基层医师对KS了解不足,往往在儿童中诊断率低下。NGS具有快速、经济、全面的优点,可以发现新的致病性或可能的致病性变体,对协助KS的诊疗具有一定意义,因此如在临床中遇到可疑KS的患者,可建议其完善NGS。
