溶液燃烧法合成LaMO3催化H2S分解制氢的活性研究
2022-08-25唐思扬刘长军岳海荣
徐 江,唐思扬,吴 潘,蒋 炜,刘长军,胡 强,岳海荣,2,梁 斌,2
(1.四川大学 化学工程学院,四川 成都 610065;2.四川大学 新能源与低碳技术研究院,四川 成都 610207;3.丽水学院 生态学院,浙江 丽水 323000)
当前全球仅炼油厂与天然气厂产生的H2S就超过4×107t/a,并随着高酸性油气田的开发不断增长[1]。工业上处理H2S的方式主要是通过克劳斯工艺将其转化成单质硫和水,同时回收热能[2]。而H2S直接分解的路线在回收硫磺的同时还可获得氢气,实现资源综合利用,为大规模获取绿氢资源提供了一条新途径。高温热分解[3]、催化分解[4-5]、电解[6]、等离子体分解[7]、微波催化分解[8]和光解[9]等方法都能将H2S分解为单质硫和氢气,但大多数方法在工业化方面还不成熟。而H2S催化分解由于操作简单,设备要求和工艺条件更贴合实际生产过程,是最具应用前景的H2S制氢技术。H2S分解受反应热力学平衡[10]和动力学的限制[11],开发高效分解催化剂是提高H2S转化率和氢气收率的关键。
Co、Cr、Cu、Mo、Ni和V等过渡金属的氧化物都具备一定的H2S催化分解活性[12-15]。然而,单金属氧化物催化剂稳定性较差,反应后易硫化失活。如何在保持催化活性的同时,提升催化剂的稳定性成为重要的研究课题。ABX3型钙钛矿复合氧化物,特别是LaMO3钙钛矿,由于具有良好的结构稳定性和催化活性,在中温H2S固体氧化物燃料电池(SOFC)[16]和含硫烃类燃料自热重整[17-18]等领域受到了广泛研究。钙钛矿材料中A位为稀土金属阳离子,B位为其他过渡金属,X为阴离子(一般为O),其中B位元素对钙钛矿材料的化学和物理性质影响显著[19]。GULDAL等[4,20]制备了LaMO3(M=Ce、Co、Cr、Cu、Mo、Sr或V)钙钛矿催化剂,在650~800 °C表现出良好的H2S催化分解活性。但该研究采用柠檬酸溶胶法制备催化剂,存在周期长、能耗高,且所得催化剂比表面积较小(≤10 m2/g)等缺点,不利于催化剂的规模化应用。
溶液燃烧合成法利用反应物之间的化学反应热来完成材料的合成过程,耗时短、能耗低,可以制备晶粒细小、比表面积大的氧化物材料[21-25]。本文采用溶液燃烧合成法,制备了钙钛矿催化剂LaMO3(M=Ce、Ni、Co、Mo、Cu、Sr、V或Cr)。通过改变B位元素,对催化剂形貌和结构进行调变,并将其用于H2S催化分解反应考察催化活性。
1 实验部分
1.1 实验试剂
La(NO3)3·6H2O(99.99%),购自上海麦克林生化科技有限公司;Cr(NO3)3·9H2O(分析纯)、(NH4)6Mo7O24(>98%)、Ce(NO3)3·6H2O(99.99%)和CO(NH2)2(分析纯),购自上海泰坦科技股份有限公司;NH4VO3、Sr(NO3)2、Ni(NO3)2·6H2O、Cu(NO3)2·3H2O和Co(NO3)2·6H2O,均为分析纯,购自成都市科隆化学品有限公司。
1.2 催化剂的合成
溶液燃烧法合成LaMO3(M=Ce、Ni、Co、Mo、Cu、Sr、V或Cr)的操作步骤完全相同,物料质量比则通过不同原料燃烧反应的化学计量关系分别计算确定。
以LaCeO3的制备为例,根据推进化学的热化学理论[26]和所选用的原料组成,按照燃烧反应的化学计量比,计算出各组分质量为:La(NO3)3·6H2O 1.73 g、Ce(NO3)3·6H2O 1.74 g以及尿素2.40 g。首先称取1.73 g的La(NO3)3·6H2O和1.74 g的Ce(NO3)3·6H2O溶于10 mL去离子水中,在80 °C的恒温水浴中搅拌混合,再称取2.40 g尿素,溶于10 mL去离子水中,随后将尿素溶液滴加到上述硝酸盐溶液中,继续加热搅拌1 h,再转移至已预热至500 °C的马弗炉中进行燃烧反应,待冷却至室温后取出,即得LaMO3前驱体粉末,见图1,充分研磨后在管式炉中以5 °C/min升温至1000 °C,煅烧5 h即得LaCeO3。

图1 LaCeO3前驱体粉末Fig.1 Precursor powder of LaCeO3
1.3 催化剂的表征
X射线衍射(XRD)测试,采用荷兰帕纳科公司Bruker D8 ADVANCE型X射线衍射仪。以Cu靶为辐射线源(λ=0.15418 nm),管电压40 kV,管电流40 mA,步长0.02°,扫描速率5 (°)/min,扫描范围为10°~80°。
催化剂孔隙结构参数测定,采用美国Micromeritics公司ASAP 2460型全自动比表面积与孔隙分析仪。先将催化剂在250 °C下进行5 h的真空脱附处理,然后在液氮温度(-196 °C)下进行N2吸附测定,利用BET方程计算催化剂比表面积。
催化剂微观形貌和元素分布观测,采用日本电子株式会社JSM 7610F扫描电子显微镜(SEM)。在不同放大倍数下观测样品形貌,采用EDS面扫描对催化剂元素的含量和分布进行测试。
1.4 催化剂的评价
催化剂催化H2S热分解反应活性评价在固定床反应器中进行,反应装置示意如图2。反应气组成为:φ(H2S) =5%,平衡气为N2,反应空速(GHSV)为48000 h-1,测试温度为600~800 °C。首先将约0.13 g 40~60目的催化剂装入内径为4 mm的石英管反应器,在N2气氛下,以10 °C/min将反应器温度升至800 °C,然后切换为反应气进行催化活性测试。反应生成的硫蒸气经反应器出口尾部石英管冷凝后固化,气相产物通过在线气相色谱(SC-3000B,TCD检测器,Porapak Q填充柱,载气为N2,重庆川仪自动化股份有限公司)进行分析。当各温度测试点、反应器出口H2S浓度达到稳定,以此时的转化数据作为催化剂活性评价基础,尾气经碱液吸收瓶净化后排空。
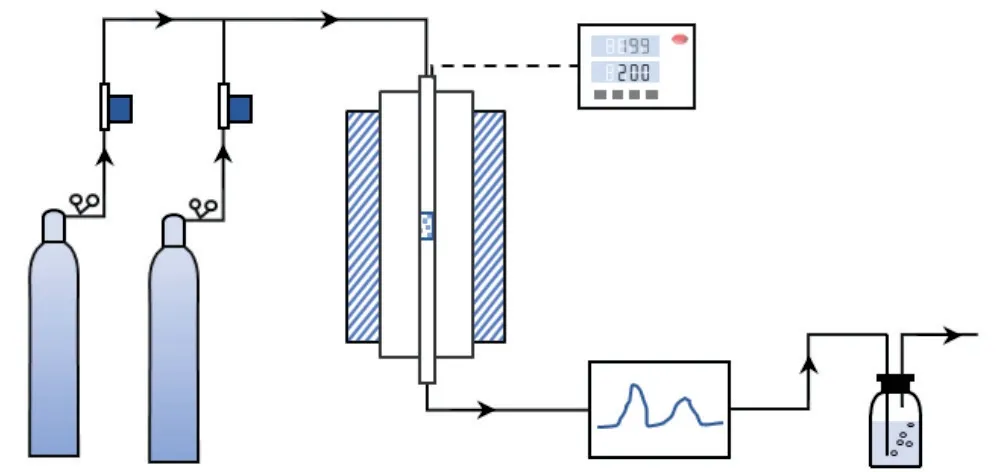
图2 催化剂活性评价装置Fig.2 Catalyst activity evaluation device
H2S转化率、H2选择性分别由式(1)、式(2)计算得到。
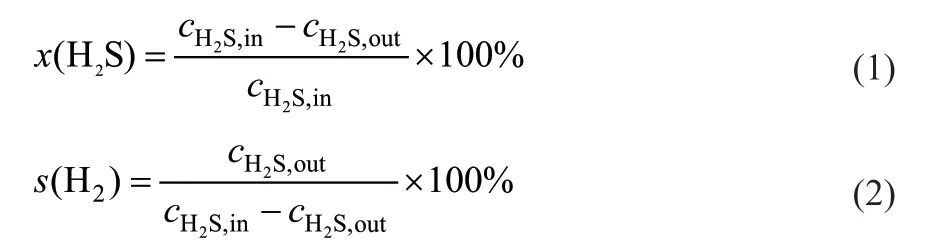
式中,c为各物质的物质的量浓度,mol/L。
2 结果与讨论
2.1 催化剂活性
通过固定床反应器对8种不同B位元素的催化剂在600~800 °C下的催化活性进行了评价,结果如图3所示。由图3(a)可知,各催化剂的催化活性都随着反应温度的升高而显著上升。在800 °C下,各催化剂的H2S转化率按Mo<Sr<V<Cr<Cu<Co<Ce<Ni的顺序依次增大,其中LaNiO3最高,达到18.2%。同时,在600~800 °C内,LaNiO3的H2S转化率都保持在较高水平,LaCrO3次之,LaCeO3、LaVO3、LaSrO3和LaMoO3相对较低。由图3(b)可知,结合H2选择性,H2S除了发生分解反应,还发生了其他副反应。在800 °C下,各催化剂H2选择性按Cr>Ce>Co>Ni>Cu>V>Sr>Mo的顺序依次减弱,其中LaCrO3最高,可达61.3%。随着反应温度的下降,大部分催化剂的H2选择性都呈现减小的趋势,而LaMoO3低温下的H2选择性则呈现出增大的趋势,这可能与LaMoO3在硫化过程中形成的MoS2有关,MoS2是催化H2S热分解的有效活性组分[14]。
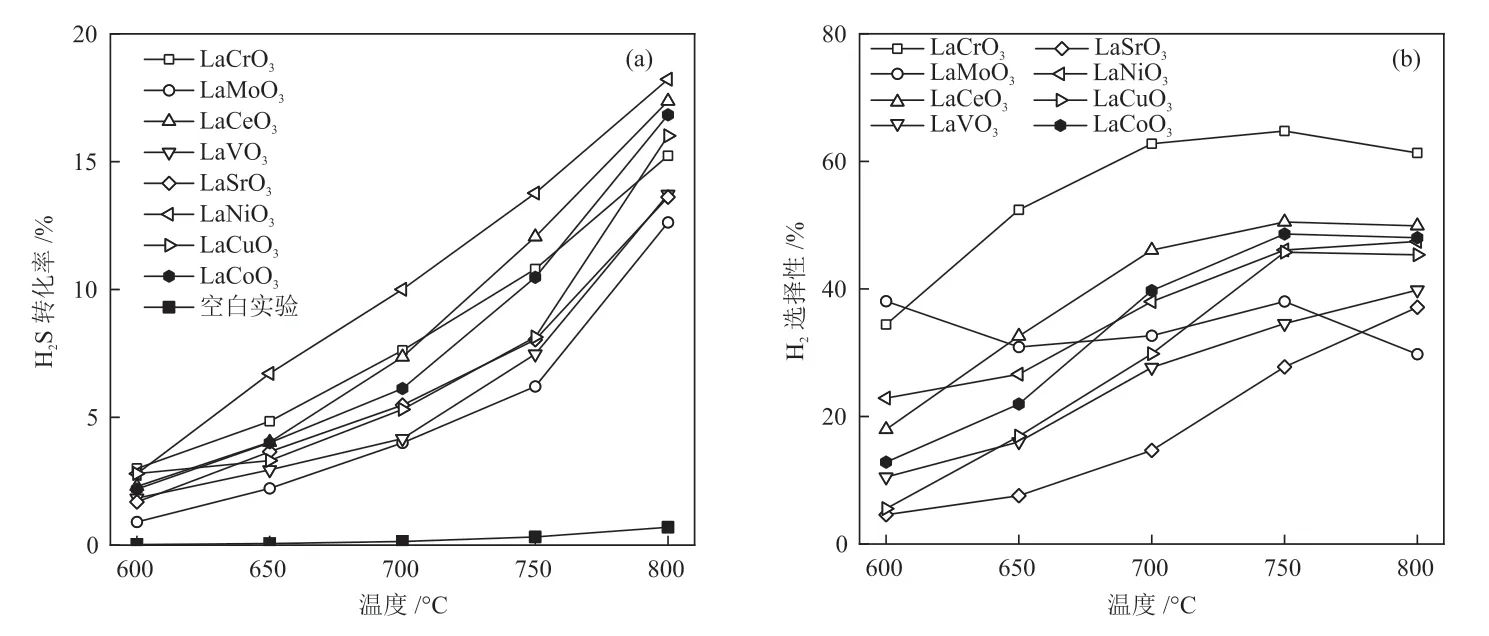
图3 反应温度对催化剂H2S转化率(a)和H2选择性(b)的影响Fig.3 Effect of reaction temperature on H2S conversion (a) and H2selectivity (b) of catalysts
2.2 催化剂孔结构特性
反应前各催化剂的BET比表面积和孔隙大小如表1所示。由表1可知,催化剂比表面积分布在0.75~14.85 m2/g。其中,LaCeO3拥有最大的比表面积和孔容,分别为14.85 m2/g和47.7×10-3cm3/g;而LaCoO3的比表面积和孔容最小,分别为0.75 m2/g和2.2×10-3cm3/g。相比之下,LaCeO3催化剂的H2S转化率和H2选择性都较好。
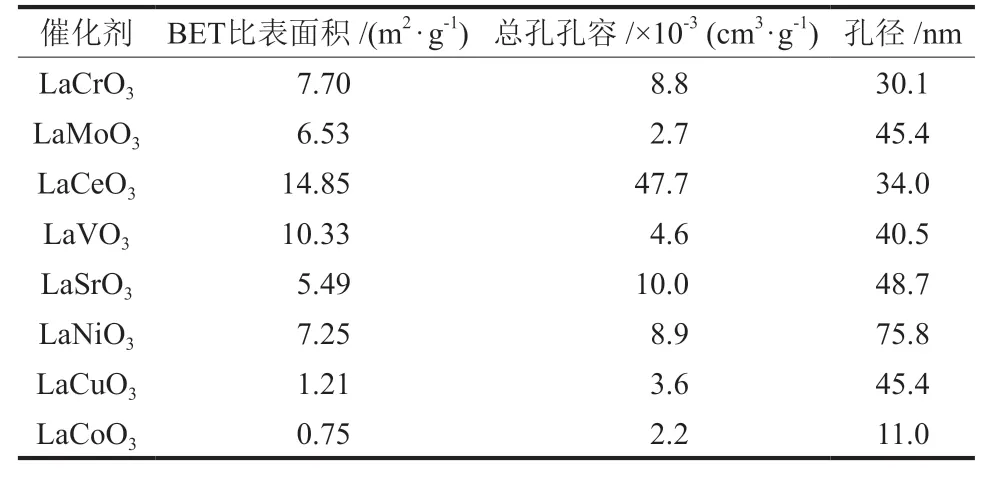
表1 反应前催化剂的BET比表面积及孔结构参数Table 1 BET specific surface area and pore structure parameters of catalysts before reaction
2.3 催化剂晶体结构
利用XRD对反应前后催化剂粉末的晶体结构进行了探究,结果如图4所示。由图4可知,反应前催化剂的XRD结果显示,采用溶液燃烧合成法制得 的LaCrO3、La2Mo2O9、LaVO4、La4Ni3O10、La2CuO4和LaCoO3具备纯相类钙钛矿结构。而反应后催化剂的XRD结果表明,所有催化剂中都有含硫新晶相的形成,这是由于部分H2S与催化剂发生硫化副反应所致。
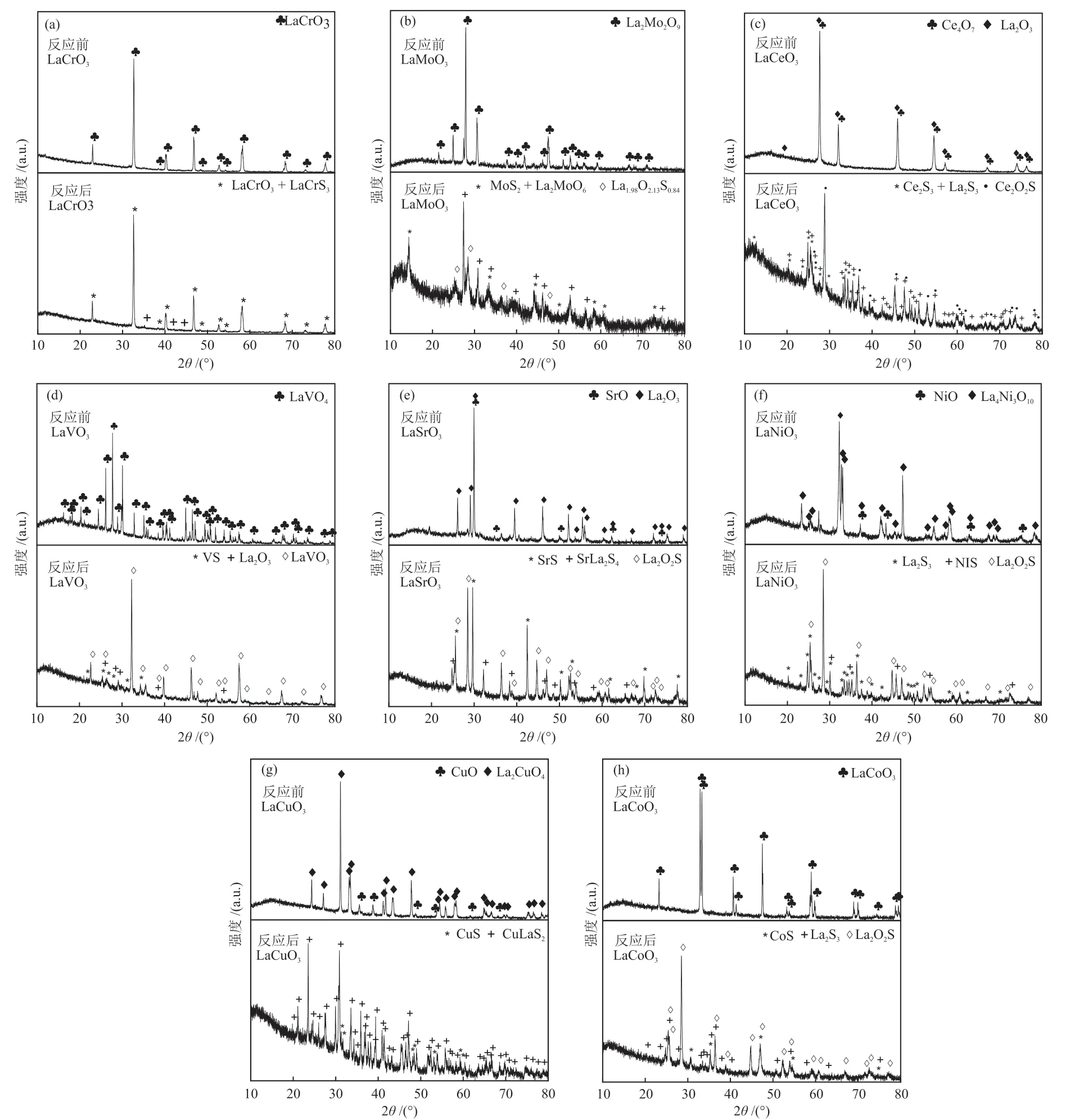
图4 反应前后催化剂的XRD谱图Fig.4 XRD patterns of catalysts before and after reaction
由图4的XRD图谱分析各催化剂的晶像组成可以看出,部分催化剂偏离了理想的ABO3钙钛矿结构,这是因为在煅烧过程中,氧气压力和温度控制着金属的氧化状态,并产生氧空位来补偿电荷中性[27-28]。在大多数反应前的样品中,LaCrO3(No.83—1327)、La2Mo2O9(No.23—1145)、LaVO4(No.70—2392)、La4Ni3O10(No.50—0243)、La2CuO4(No.82—2139)和LaCoO3(No.48—0123)单钙钛矿相明显。对含Ce和Sr样品的分析表明,只有相应的单金属氧化物相,没有显示任何钙钛矿相。这可能是由于在材料制备过程中,空气气氛常压下,煅烧前驱体粉末阶段发生了氧化铈和氧化镧相的偏析,关于钙钛矿结构中Ce的相似相和有限溶解度在文献中已有报道[29]。与之类似,氧化锶和氧化镧相也存在这种偏析现象[30]。因此,在含Ce和Sr样品中没有观察到明显的钙钛矿相。
在反应后的样品中,只有LaCrO3催化剂很好地保持了原有的钙钛矿晶相LaCrO3(No.83—1327),并有少量硫原子取代了氧,形成了新相LaCrS3(No.89—8650)。LaMoO3和LaVO3催化剂反应后的钙钛矿晶型发生了变化,形成了新相La2MoO6(No.24—0550)和LaVO3(No.78—2305),并 出 现了新的含硫相MoS2(No.24—0513)、La1.98O2.13S0.84(No.89—6895)和VS(No.73—2024)。而LaNiO3、LaCuO3和LaCoO3催化剂,在反应后已观察不到反应前的钙钛矿相,只有新的含硫相La2S3(No.22—0645)、NiS(No.65-3419)、La2O2S(No.71—2098)、CuLaS2(No.73—1858)、CuS(No.65—7111)和CoS(No.65—3418)存在。反应后的LaCeO3和LaSrO3催化剂同样只存在含硫新相Ce2S3(No.20—0269)、La2S3(No.22—0645)、Ce2O2S(No.24—0289)、SrS(No.65—9581)、SrLa2S4(No.29—1307)和La2O2S(No.71—2098)。
2.4 催化剂元素组成及其形貌
反应前后催化剂的EDS元素组成分析如表2所示。反应后的催化剂中含有部分硫原子,部分氧原子被硫取代,催化剂被硫化。通过元素分析考察了B位元素对催化剂硫化程度的影响,各催化剂硫化程度依照Cr<V=Ce<Sr<Cu<Co<Mo<Ni顺序增大。B位元素直接影响钙钛矿材料的物理化学性质,不同B位掺杂催化剂的硫化程度不同,含Ni催化剂最高,而含Cr催化剂最低。由XRD图谱(图4)也能看出,反应后LaCrO3催化剂的钙钛矿晶型保持良好,其耐硫性最好。LaNiO3虽然在所有催化剂中具有最高的H2S转化率,但其反应后的硫化程度最高,H2选择性差。LaCrO3的H2S转化率并不突出,但其反应后的催化剂硫化程度不高,很好地保持了原有的钙钛矿结构,有利于产氢活性的表达,因此其H2选择性最好,产氢性能最优。LaNiO3较高的硫化程度可能与Ni对硫的敏感度有关,用于甲烷重整的Ni基催化剂,往往因极低浓度的含硫燃料,发生硫中毒而迅速失活[31]。
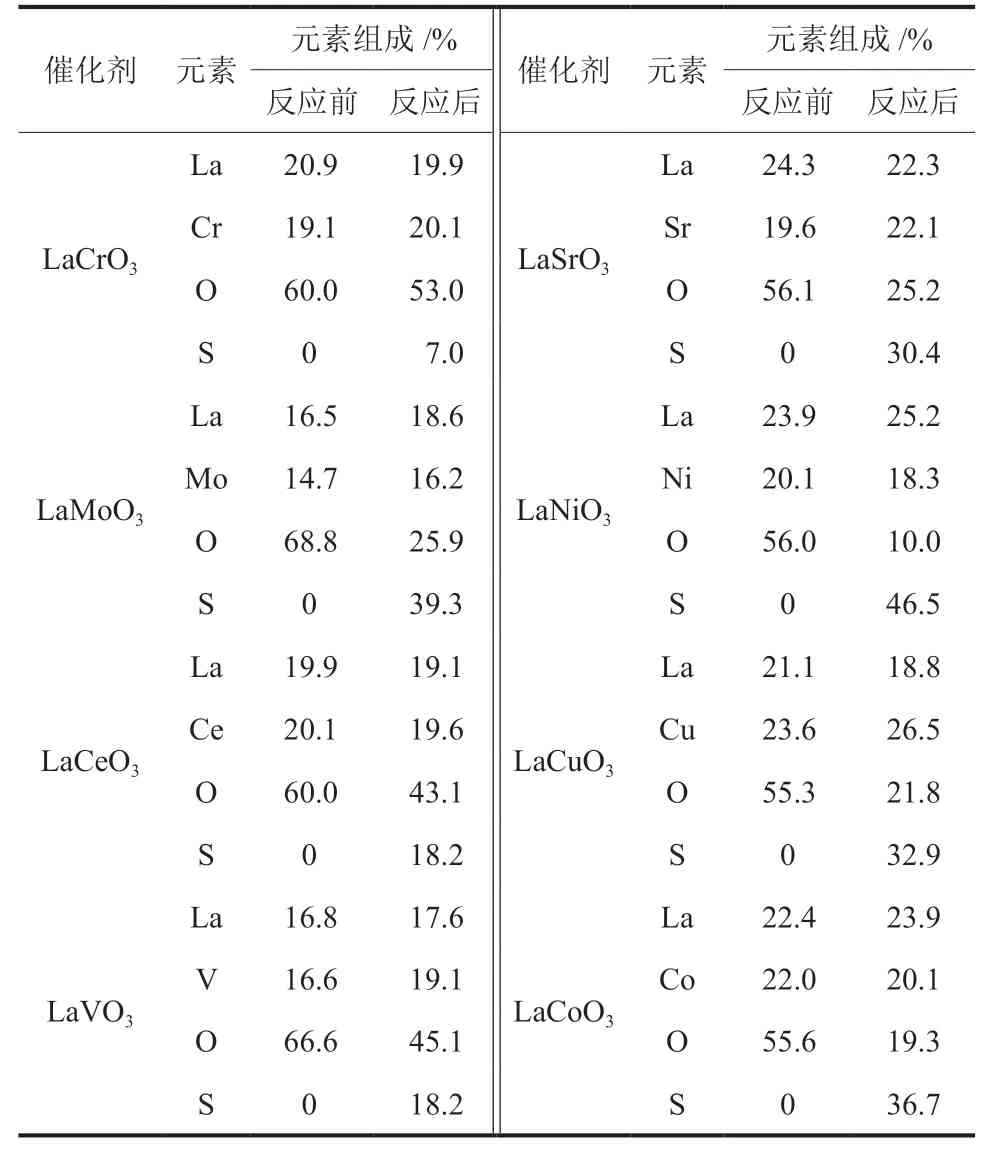
表2 反应前后催化剂的EDS元素组成Table 2 Elemental composition of catalysts before and after reaction by EDS
采用SEM对反应前后催化剂的微观结构进行了分析,结果如图5所示。由图5可知,反应前后各催化剂的晶粒尺寸没有发生明显变化,表明反应过程中催化剂颗粒无明显团聚。反应后催化剂表面相对变得粗糙,这是由于催化剂表面颗粒被硫化造成的。如图5(c)和(d)中,反应后LaMoO3催化剂表面明显变粗糙,且有片状结构物质生成,这可能是在反应过程中催化剂被硫化,生成了MoS2,在XRD谱图(图4)中也观察到该物质的生成。结合比表面积数据(表1)可知,LaCeO3催化剂颗粒粒径最小,且存在部分孔道结构,其比表面积和孔容较大;而LaCoO3和LaCuO3等催化剂的颗粒粒径较大,且没有明显孔道结构,其比表面积和孔容较小。
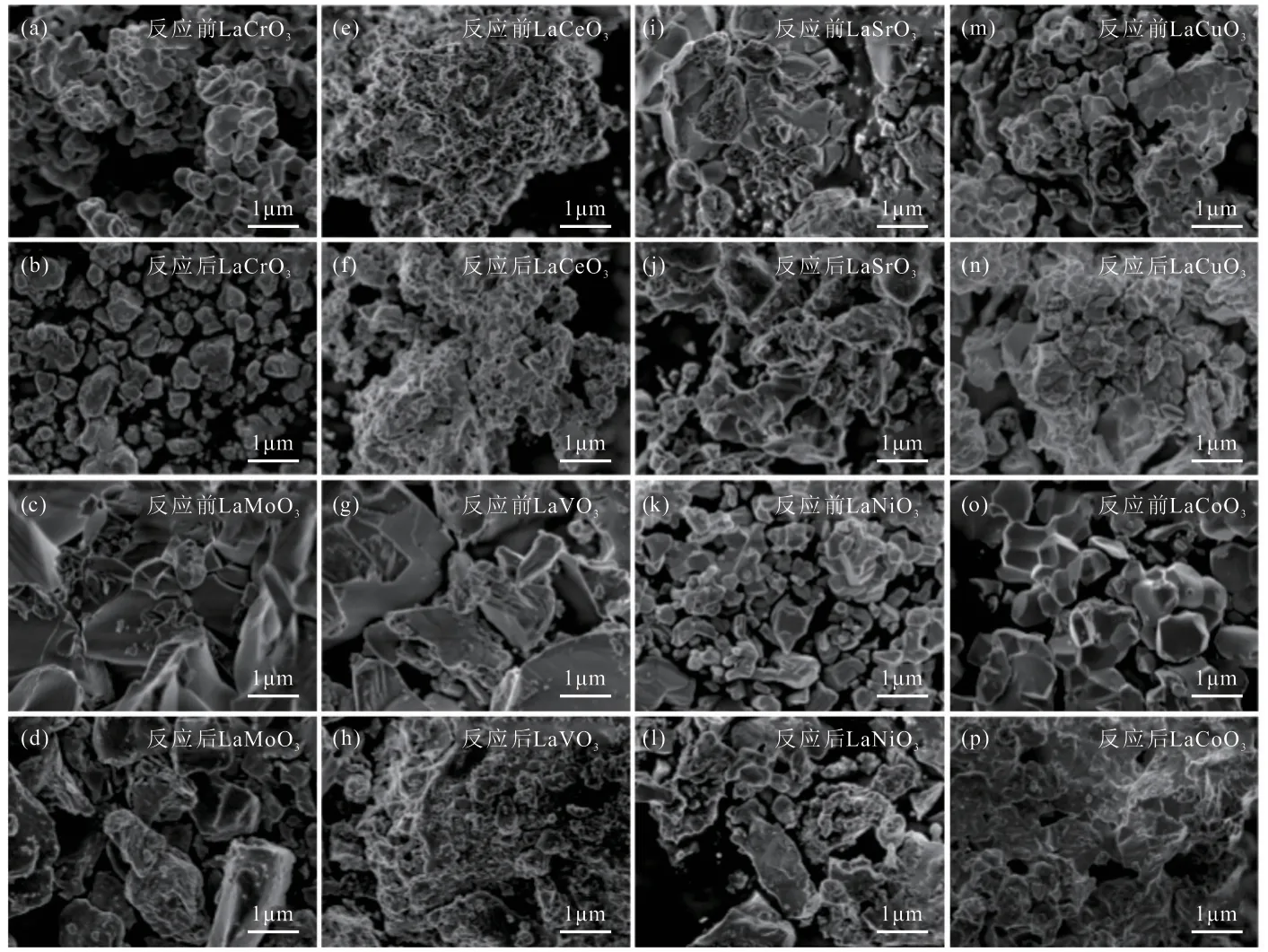
图5 反应前后催化剂的SEM照片Fig.5 SEM images of catalysts before and after reaction
3 结论
本文采用溶液燃烧合成法制备了钙钛矿催化剂LaMO3,评价了其H2S催化分解制氢的活性,探究了不同B位掺杂元素(Ce、Ni、Co、Mo、Cu、Sr、V和Cr)对催化剂物理化学性质的影响,得到如下结论。
(1)在600~800 °C,GHSV为48000 h-1的反应条件下,8种LaMO3钙钛矿材料对H2S分解均具有催化作用。其中LaCrO3的H2选择性最高,在800 °C时可达61.3%;LaNiO3的H2S转化率最高,在800 °C时可达18.2%,但H2选择性差。相比下,LaCrO3具有最好的产氢活性。
(2)采用溶液燃烧合成法,可制得较大比表面积的催化剂材料。其中,LaCeO3的BET比表面积和孔容最大,分别可达14.85 m2/g和47.7×10-3cm3/g。H2S催化分解过程中,催化剂表面发生了硫化反应并生成了新的硫化物相。催化剂硫化程度按Cr<V=Ce<Sr<Cu<Co<Mo<Ni依次增大,LaCrO3硫化程度最低。
