7例新生儿皮肤异常的临床特征及遗传分析
2022-08-25戴立英刘光辉
王 影 戴立英 刘光辉
色素失禁症( incontinentia pigmenti, IP)是一种罕见的、由IKBKG基因杂合性致病变异导致的X连锁遗传病,男性患病胎儿多因IKBKG变异半合子在出生前夭折,仅少数因体细胞嵌合体或XXY异常核型(即Klinefelter综合征)而得以存活[1];女性患者则多数因X染色体重排导致的杂合性IKBKG4~10号外显子缺失致病;全球发病率约1/143,000,男女发病比率约为1∶20[2](ORPHA:464, Orphanet)。IP本质上是一种外胚层发育不良病[3],因此IP患者多见外胚层来源的皮肤损害和牙齿、头发、指甲和视网膜异常。然而在新生儿期(不足1个月龄),患儿相关器官发育尚不完全,且不能配合进一步检查,多数情况仅能通过皮损特征以及家族史等有限信息来考虑IP诊断,因此诊断和鉴别诊断较为困难。
针对IKBKG片段缺失和点突变的单基因检测是目前常用的IP诊断方法,但在新生儿患者中,该方法阳性率及鉴别诊断的效能则可能存在疑问。对此,本研究对7例新生儿期疑似IP患儿进行了遗传性病因的探讨,并通过对此罕见病新生儿表型的总结,以期提高新生儿期IP的临床诊断水平。
1 资料与方法
1.1 一般资料 回顾性分析2019年6月至2021年6月医院收治的7例疑似IP诊断的新生儿(均为1个月龄内)。
1.2 方法
1.2.1 新生儿IP的临床诊断标准 在Landy和Donnai提出的IP诊断标准基础上,遵循简化的新生儿期诊断标准:①皮疹、红斑、水疱、典型的线状色素沉着等皮肤异常,(条件允许的话)且皮肤样本有嗜酸细胞小囊泡的病理学改变;②其他外胚层异常,羊毛样卷发,指甲异常等,则支持IP诊断;③直系女性IP家族史,或多次妊娠男胎流产史,则支持IP诊断。
1.2.2 临床特征收集 除患儿一般信息,根据人类表型本体中文版数据库标准对患儿的临床表型进行总结,并与美国国家健康研究所的遗传病及罕见病信息中心提供的IP临床表型(https://rarediseases.info.nih.gov/diseases/6778/incontinentia-pigmenti)进行对比,用以比较所发现表型在以往报导IP患者中的发生率。
1.2.3 基因检测 所有疑似IP新生儿均使用PCR(gap-PCR,高敏感性多重PCR),引物设计见图1[4],Sanger测序进行外周血检测,分别对热点的4~10外显子缺失和点突变进行检测。对于gap-PCR+Sanger测序阴性患者,续而通过加深全外显子组测序(whole exome sequencing,WES)进一步寻找遗传性病因。WES文库构建采用xGen© Exome Research Panel v1.0捕获探针,通过NovaSeq 6000(Illumina,美国)测序仪进行双端短读长测序(PE150),100X测序深度时,目标序列测序覆盖度不低于99%,而在本研究中,WES测序深度为300X,从而能更好地检出体细胞嵌合突变。Gap-PCR+Sanger测序检测和WES均由智因东方转化医学研究中心有限公司(Chigene,北京)完成,其中WES分析在智因东方遗传病分析云平台(https://cloud.chigene.org)上进行。最后,通过WES鉴定的基因变异均通过Sanger测序进行验证。
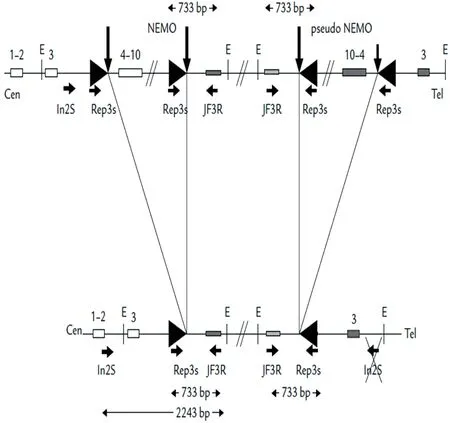
图1 检测IKBKG热点重排导致外显子4-10
2 结果
2.1 一般资料 7例色素失禁症患儿中,均为女性,其中1例早产,出生胎龄35周,7例中4例剖宫产出生,3例顺产出生,出生体质量2 200~3 240 g; 其父母亲均体健,孕期无任何疾病及服药史,也否认冶游史,宫内感染系列(弓形虫、巨细胞病毒、单纯疱疹病毒 Ⅰ及单纯疱疹病毒Ⅱ) 检查均为阴性,梅毒抗体检测也为阴性。所有患儿均有皮疹及嗜酸性粒细胞增多,7例患儿中头颅MR异常2例,眼底异常改变2例;其中2例有家族高位病史,外祖母属近亲婚配,有脑性瘫痪及皮肤病患者。
2.2 临床表现 新生儿期的临床表现主要为皮疹,其中5例以皮疹为主诉收住院,皮疹特点: 出生时即有皮疹6例,生后第3天出疹1例。皮疹分布: 以四肢内侧及躯干多见,呈线性分布;确诊IP患儿的皮疹多伴有皮屑,可伴有红斑及色素沉着。见图2。1例患儿有肺部感染,伴有多次不对称的肢体痉挛发作且伴有头颅MR异常及眼底改变;1例患儿心肌酶异常升高,提示心肌损害。
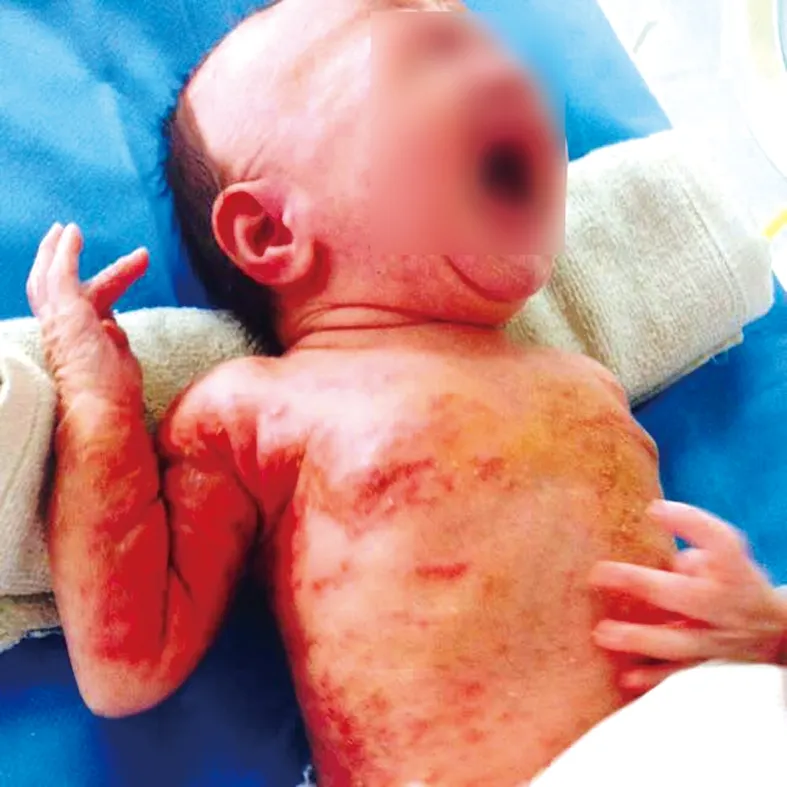
图2 基因检测确诊为新生儿IP
2.3 血常规检查 所有患儿血常规嗜酸性粒细胞计数(0.21~6.18)×109,均呈不同程度升高。
2.4 基因检测 7例患儿均经gap-PCR+Sanger测序,其中5例经基因检测确诊为IP,2例为4~10号外显子缺失,3例为IKBKG点突变(图3),分别为NM_001099856: c.358C>T,p.Q120*、NM_003639:c.831delA,p.K277Nfs*4、NM_001099857:exon2:c.184C>T,p.R62*,这3个点突变均为截断突变,可导致编码蛋白提前终止,另外这3个变异在国内外正常人群体携带频率极低(参考GnomAD/dpSNP/1000G/ESP6500数据库),根据ACMG指南变异评级,均符合致病性或可能致病性变异。其中,p.Q120*、p.K277Nfs*4目前均已被研究[5-6]报道过,而p.K277Nfs*4在HGMD/Clinvar等已知致病变异数据库均未见收录,属于未报导过的新变异。gap-PCR阴性结果的患者中,1例后续经WES检测发现携带PLEC基因复合杂合性变异c.6173G>A和c.2846T>C,见图4,该患儿的临床表型与遗传学方式均符合PLEC基因变异相关所致大疱性表皮松解症;另1例因拒绝进一步WES检测。
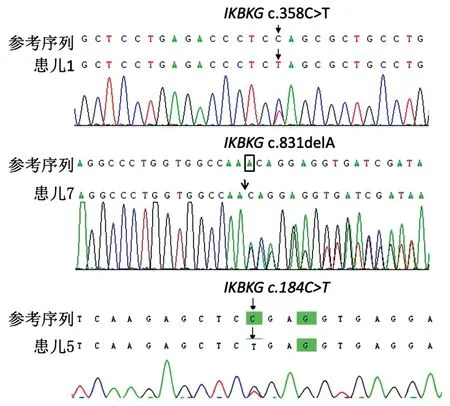
图3 3例患者携带IKBKG点突变,分别为NM_001099856:

图4 Sanger测序验证,患者携带PLEC基因c.6173G>A杂合突变,遗传自母亲,父亲为野生型;携带PLEC基因c.2846G>A杂合突变,遗传自父亲,母亲为野生型
2.5 治疗与预后 本病无特殊治疗,主要加强皮肤护理,预防感染。通常在2岁以后逐渐消退,到成年期除一些原有的并发症外,无其他不适。在水泡期应注意防止继发感染,可外用含肾上腺皮质激素类抗生素软膏。
3 讨论
IP是一种罕见的X连锁显性遗传性疾病,临床表现有特征性皮肤改变,可伴眼、骨骼和中枢神经系统畸形和异常,男女发病比例为1∶20。男性患儿通常在宫内死亡,多数存活的IP男性患儿多由NEMO基因的点突变导致。因此临床上绝大多数为女性患儿,女性患儿常在出生时或出生后不久发病,以皮肤损害为特征,除皮肤损害外,还可能出现脱发、牙齿发育不全或先天性无牙、甲发育异常、角膜病变、白内障、视网膜血管异常增生以及视网膜剥离等。此外,神经系统并发症是另一突出表现。临床上较常见新生儿时期头颅MR及临床特征诊断脑损伤。脑损伤会导致神经系统后遗症,如智力障碍、脑瘫等[7-8]。本文搜集的7例患儿均为女性,其皮肤损害的程度和范围个体差异性很大,IP患儿皮疹特点新旧重叠出现。囊泡期的皮疹可以不存在或发生在宫内,这种皮肤损害大约在4个月龄时消失,部分皮疹通常在2岁以后逐渐消退,到成年期除一些原有的并发症外,无其它不适。本病对于皮肤损害尚无特异性治疗方案,主要是避免皮肤感染。该病远期预后取决于皮肤外其他系统受累情况,尤其是神经系统和眼部受累的治疗相对困难,可能预后不良。
本研究通过突变筛查,目前已明确IP的NEMO基因缺失突变(第4~10外显子缺失)是80% IP发病的原因[9]。除了最常见的NEMO基因第4~10外显子大片段缺失外,还有假基因△NEMO共有序列缺失[10-11]。在已经发现基因突变类型中,90%为假基因△NEMO中共有序列NEMOΔ4-10缺失,仅10%明确为微小突变[12-13]。本研究中进行基因检测的7例患儿中有5例检测出NEMO 第4~10外显子缺失,有2例检测出NEMO基因点突变。
本研究中所有IP患儿均在新生儿时期发病,且多以皮疹为首发表现,这与国内外的研究一致[14-15]。由于患儿就诊时间均在新生儿期,所以皮疹均为第1、2期改变。IP为X连锁显性遗传病,由位于染色体Xq28上的NEMO基因突变所引起。IP患儿生后由于上皮细胞内含NEMO突变的角质细胞,生成大量的白细胞介素-1β,诱导肿瘤坏死因子-α合成,肿瘤坏死因子-α促使突变的角质细胞凋亡,触发皮肤的炎性反应[16],发生皮肤损伤。目前约80%的IP病例可以找到基因突变,另有20%尚未明确病因。而临床表型轻微,并证实未发现NEMO基因内共有序列NEMOΔ4-10缺失,可进一步检测微小突变。NEMO基因突变分析可证实临床诊断,并可提供产前诊断技术,值得应用和推广。本研究结果进一步证明我国绝大多数IP基因改变类型为共有序列NEMOΔ4-10缺失,为产前诊断的开展提供一定依据。
