先天性非溶血性高间接胆红素血症19例临床分析*
2022-08-22高丹田昌军刘芳平覃大卫张伟田文军谷秀
高丹 田昌军 刘芳平 覃大卫 张伟 田文军 谷秀
先天性的胆红素-尿苷二磷酸葡萄糖醛酸转移酶(bilirubin-uridine diphosphate glucuronosyl- transferase,UGT1A1)缺陷或活性低下导致血清中间接胆红素的升高,称之为先天性非溶血性高间接胆红素血症。其发病的遗传基础是位于染色体2q37位点上UGT1A1基因发生突变,主要表现为常染色体隐性遗传,根据此酶缺乏程度和基因分析的不同,可分为 Gilbert综合征和Crigler-Najjar综合征I型和Ⅱ型[1]。先天性非溶血性高间接胆红素血症患儿可以出现与血清胆红素升高相关的临床表现,此类疾病新生儿期常表现为皮肤重度黄染,若体内胆红素水平持续重度升高可对患儿的脑部组织产生损伤,引起胆红素脑病等,从而影响新生儿的运动功能、智力等的发育,甚至可造成其死亡[2]。国内外的研究已发现该病有明显的遗传异质性,约有40余个UGT1A1基因突变与该病相关,并且突变谱存在明显的地区和人群差异[3]。关于本地区先天性非溶血性高间接胆红素血症新生儿的UGT1A1基因突变情况以及该类疾病患者的临床特点、治疗预后研究较为少见,鉴于此,现将我院收治的19例先天性非溶血性高间接胆红素血症病例进行回顾性分析。
对象与方法
1 研究对象 纳入标准:2019年1月~2021年3月某医院新生儿科根据《实用新生儿学》(第5版)的先天性非溶血性高间接胆红素血症诊断及分类标准[1],通过全外显子测序明确诊断先天性非溶血性高间接胆红素血症的足月新生儿。排除标准:低出生体重儿、早产、先天畸形、母婴血型不合导致的溶血、头皮血肿、新生儿败血症、G-6-PD缺乏、红细胞增多症等影响黄疸程度的因素;合并脑积水、脑回畸形等颅脑发育异常;影响智力发育的遗传代谢病及染色体病。
2 研究方法 收集患儿性别、起病日龄、入院日龄、确诊时间、住院天数、临床表现及临床治疗情况。通过实验室及影像学检查分析患儿血清胆红素水平及脑功能情况。通过电话及网络问卷等方式进行随访,平均随访时间1年,主要包括生存情况、辅助检查情况、接受治疗情况、原发病缓解情况(原发病缓解指住院期间的临床症状减轻或消失,未再出现新的合并症或新的合并症减轻或消失、不需要行酶诱导剂及光疗等治疗)。
3 统计学方法 采用SPSS 23.0软件对数据进行统计学分析。所有数据进行正态性检验,符合正态分布的计量资料用(±s)表示,使用t检验,当P<0.05时则提示差异存在统计学意义;不符合正态分布的计量资料采用中位数(四分位数间距)表示。用百分号(%)表示计数资料。
结果
1 一般资料与临床特征 19例足月UGT1A1基因突变患儿中,男12例,女7例,汉族2例,土家族17例,平均胎龄(272.6±8.9)d,平均出生体重(3340.0±35.5)g,起病时间中位数2 d(四分位数间距:1,3 d),中位入院时日龄7 d(四分位数间距:5,9 d),中位住院天数7 d(四分位数间距:6,9 d),中位确诊时间 32.5 d(四分位数间距:30,34 d),平均总胆红素值为(374.9±91.7)μmol/L,平均光疗时间为(37.0±15.3)h。
2 基因测序结果分析 通过二代测序技术进行全外显子检测,确定UGT1A1基因位点突变患儿19例,共6个位点:Exon1 c.211G>A(p.(Gly71Arg))突变16例,Exon4 c.1091C>T(p.(Pro364Leu))突变3例,Exon5 c.1318A>G(p.(Ile440Val))突变1例,Exon1 c.1348C>T(p.(Arg450Cys))突变1例,Exon1 c.686C>A (p.(Pro229Gln))突变1例,Exon5 c.1456T>G (p.(Tyr486Asp))突变1例。其中单纯纯合突变最多共10例(52.63%)、单纯杂合突变5例(26.32%)及复合杂合突变4例(21.05%)。19例患儿基因突变类型均为错义突变。详见表1及图1。

图1 UGT1A1基因突变位点(正向测序)

表1 19例UGT1A1突变患儿的基因测序结果分析
3 临床表现及辅助检查 19例患儿临床表现中,皮肤巩膜重度黄染14例、惊跳11例、易激惹2例、肌张力高3例、精神反应欠佳5例、肢体抖动2例;辅助检查中,重度高胆红素血症7例(TSB>342 mmol/L)、极重度高胆红素血症(TSB>427 mmol/L)6例、危险性高胆红素血症(TSB>510 mmol/L)1例,共14例,乳酸中毒3例、心肌损害5例、呼吸性碱中毒6例,基底节区苍白球T1WI信号增高6例,24 h动态脑电图异常5例,见图2。

图2 19例先天性非溶血性高间接胆红素血症患儿临床表现及辅助检查特征
4 治疗及随访结果 19例患儿中,7例患儿同时接受外周动静脉同步换血及间歇性双面蓝光疗法,接受换血疗法中位时间为生后7.2天,接受光疗时间平均(34.7±17.0)h。12例患儿行间歇性双面蓝光疗法,接受光疗时间平均(38.3±12.7)h,所有患儿同时接受苯巴比妥5 g诱导酶活性治疗,中位使用时间5 d(四分位数间距:4,6 d)。治疗后原发病症状较前缓解,出院前及出生后1月龄血清间接胆红素水平均较前下降(P<0.001,见表2),3例患儿出现胆汁淤积症,其中1例病情反复。随访发现19例患儿基本生长发育情况均尚可,其中1例危险性高胆红素血症患儿神经精神症状持续至2月龄。
表2 19例患儿治疗前后血清肝功能指标水平 (±s)
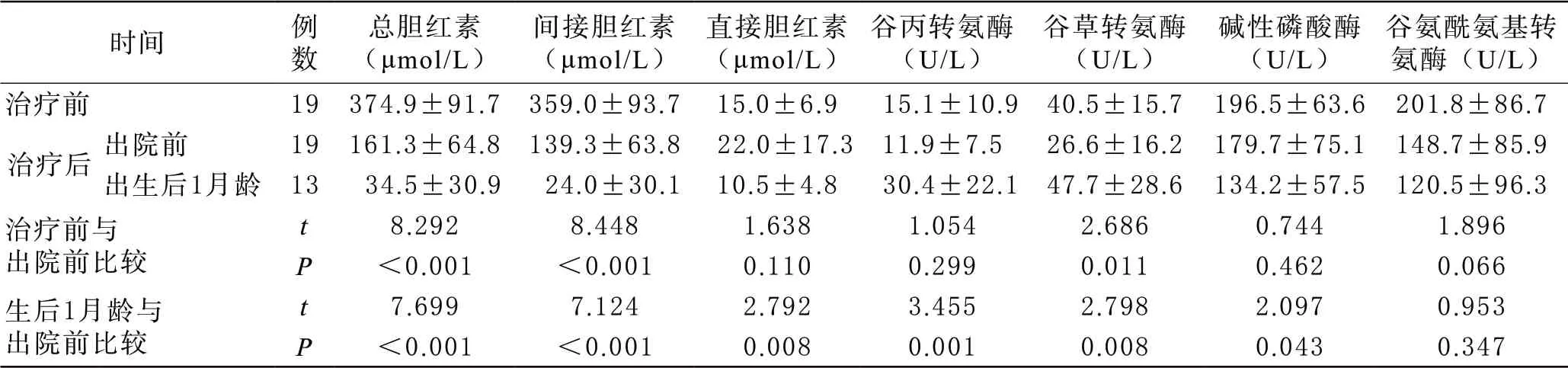
表2 19例患儿治疗前后血清肝功能指标水平 (±s)
时间 例数总胆红素(μmol/L)间接胆红素(μmol/L)直接胆红素(μmol/L)谷丙转氨酶(U/L)谷草转氨酶(U/L)碱性磷酸酶(U/L)谷氨酰氨基转氨酶(U/L)治疗前 19374.9±91.7359.0±93.715.0±6.915.1±10.940.5±15.7196.5±63.6201.8±86.7治疗后 出院前 19161.3±64.8139.3±63.822.0±17.311.9±7.526.6±16.2179.7±75.1148.7±85.9出生后1月龄 1334.5±30.924.0±30.110.5±4.830.4±22.147.7±28.6134.2±57.5120.5±96.3治疗前与出院前比较t 8.2928.4481.6381.0542.6860.7441.896 P <0.001 <0.0010.1100.2990.0110.4620.066生后1月龄与出院前比较t 7.6997.1242.7923.4552.7982.0970.953 P <0.001 <0.0010.0080.0010.0080.0430.347
讨论
近年来,新生儿不明原因黄疸发病率高达20%~30%[4],而先天性非溶血性高间接胆红素血症是其中的一个重要病因[5],对临床上诊断及治疗新生儿不明原因高胆红素血症提供了很多帮助。本组病例中可以观察到皮肤、巩膜重度黄染以及神经精神症状是先天性非溶血性高间接胆红素血症患儿最常见的临床症状。此外,患儿实验室检查以血清间接胆红素极度增高为主,共14例,这可能与UGT1A1基因突变导致酶活性减低有关,使得体内非结合胆红素转化为结合胆红素代谢发生异常,从而出现以间接胆红素升高为主的高胆红素血症[6]。通过影像学及脑电图检查发现,患儿早期可出现基底节区苍白球T1W1信号增高及24 h动态脑电图异常的表现,分别占6/5,其原因可能是胆红素通过与成熟神经元的神经节苷脂和磷脂相结合损害神经元有关,而新生儿期在生理及生化代谢方面以基底节核神经细胞最为活跃,耗氧量及能量需要均最大,故基底节区最易受损[7]。随病情进展可引起神经精神症状甚至死亡。因此,对不明原因重症高胆红素血症患儿应尽快通过基因学检查明确诊断以及早期干预治疗。二代测序技术(next generation sequences,NGS)已成为确诊先天性非溶血性高胆红素血症较普遍及可靠的方法。本研究中的病例均是通过NGS检测最终诊断。
现有研究表明UGT1A1基因突变与新生儿重症高胆红素血症的发生相关[8],且UGT1A1基因突变谱存在着人种和地域差异:欧洲、美洲等白种人以TATA启动子的插入或缺失突变多见,中国、日本、泰国等亚洲黄种人以编码区核苷酸的211位点突变多见[9]。近年国内广西、云南、新疆、重庆等地的研究也揭示了UGT1A1基因突变存在民族或地域差异[10-13]。本组19例患儿中c.211G>A位点突变达16例,其中以单纯纯合突变最多见,且均为土家族新生儿。因此,该种突变可能是张家界地区土家族先天性非溶血性高间接胆红素血症患儿中常见的类型。目前已证实c.211G>A突变可影响UGT1A1活性,其纯合和杂合突变会使UGT1A1活性分别下降60.2%和32.2%,且纯合突变型患儿血清胆红素水平显著高于杂合突变型和野生型,是新生儿重症高胆红素血症发生的重要原因[14-16]。除c.211G>A位点突变外,还发现另外5个突变位点:c.1091C>T、c.1456T>G、c.1348C>T、c.686C>A及c.1318A>G,其中4个位点合并存在c.211G>A突变,难以直接通过临床特征分析其对新生儿高胆红素血症的影响。基因功能分析显示c.1091C>T及c.1456T>G突变均为致病性,c.1318A>G突变为可疑致病性。相关研究指出c.1091C>T变异是云南丘北地区常见的UGT1A1突变类型之一[17];而c.1456T>G纯合突变在云南少数民族中常见,c.211G>A、c.1456T>G复合杂合突变致病则在云南汉族人群相对多见[18];c.1318A>G突变与新生儿高胆红素血症的相关性尚未明确。因本次病例数偏少,UGT1A1基因突变类型与新生儿高胆红素血症严重程度相关性无法得出结论,尚需进一步扩大样本量探究。
目前先天性非溶血性高间接胆红素患儿通过参照美国儿科学会(AAP)推荐的光疗标准和换血标准[7],并同时辅以酶诱导剂等治疗可以取得良好的效果。本组病例中,不论是同时接受外周动静脉同步换血及间歇性双面蓝光疗法或单独接受间歇性双面光疗患儿,治疗后血清间接胆红素水平较治疗前均显著减低(P<0.001)且病情有所改善,这与葛敏[19]等人的研究结果有相似之处。通过追踪此组患儿1月龄复查结果发现血清间接胆红素没有再次上升,结合患儿临床特征及辅助检查提示体内UGT1A1酶活性可能尚未完全丧失,考虑Crigler-Najjar综合征Ⅱ型(CNSⅡ)可能。根据CNS Ⅱ有关研究结果显示,该类患者肝细胞中UGT1A1活性是正常人的60%~90%[20],但目前临床上关于该酶活性的检测技术尚未普及,不同位点变异类型导致UGT1A1酶活性改变程度仍待进一步研究。另外,治疗过程中新发胆汁淤积症3例,其中病情反复1例,遗传代谢筛查及肝胆彩超未见异常,其原因可能是光疗后体内胆红素光异构体、卟啉以及其他代谢物的累积,但机制尚未完全清楚,通常此类胆汁淤积症可以自行恢复,但后期胆汁淤积加重引起肝功能异常可能导致胆汁淤积性肝炎[21]。通过随访发现,本组患儿中出现典型胆红素脑病神经精神症状者1例,持续至2月龄左右症状消失,目前患儿基本生长发育评估尚可,因智力、语言等发育评估需动态进行,故远期预后待进一步追踪。
总之,新生儿期起病的先天性非溶血性高间接胆红素血症患儿临床表现主要以皮肤重度黄染及神经精神症状常见,本组大部分病例血清胆红素值超出重度高胆红素血症水平(>342 mmol/L);发病可能与c.211G>A、c.1091C>T、c.1318A>G、c.1348C>T、c.686C>A、c.1456T>G突变相关,且以前两者突变常见,以隐性遗传方式传递;通过外周动静脉同步换血及间歇性双面蓝光疗法可降低本组患儿血清间接胆红素水平,是缓解原发病症状的有效手段,但在治疗及随访过程中仍需警惕新生儿胆红素脑病及胆汁淤积性肝炎等并发症。
利益冲突所有作者均声明不存在利益冲突
