粉末压片制样-X射线荧光光谱法测定二氧化锆材料中二氧化锆的含量
2022-08-16周双清徐建平李明昌
周双清 ,徐建平 ,李明昌
(1.武汉科技大学 省部共建耐火材料与冶金国家重点实验室,武汉 430081;2.安徽长江钢铁股份有限公司,马鞍山 243100)
二氧化锆因具有非常好的耐酸碱、耐腐蚀、耐高温等特性,被广泛应用在航空航天、电子、高温材料等领域。二氧化锆材料中二氧化锆含量对材料性能影响较大,因此准确、快速测定其含量具有重要意义。目前,相关测定方法有湿法[1-2]、电感耦合等离子体原子发射光谱法[3-4]、熔融制样-X 射线荧光光谱法[5-7]、粉末压片制样-X 射线荧光光谱法[8-9]等。其中,湿法存在操作繁琐、速率慢、易受铪元素干扰等缺点;电感耦合等离子体原子发射光谱法以及熔融制样-X 射线荧光光谱法都存在预处理时间长、成本高等缺点;粉末压片制样-X 射线荧光光谱法尽管制样简单、速率快、成本低,但直接压片法测定主量成分含量的准确度不高。
文献[10-12]以粉末压片制样-X 射线荧光光谱法测定三氧化二铝和镁砂中主量成分和杂质的含量,结果显示,该方法所得杂质测定值与熔融制样-X 射线荧光光谱法、湿法的差别不大,而主量成分含量只有在用差减法时才能获得较高的准确度。在采用差减法计算时,所测杂质种类越全面,误差越小,主量成分的测定结果越准确。现有方法仅关注常见杂质(如二氧化硅、三氧化二铝、三氧化二铁、氧化钙、氧化镁、二氧化钛、二氧化铪和三氧化二钇等),存在着非常见成分(如三氧化钼、三氧化二钕和二氧化铈等)带来的正干扰。除此之外,粉末压片法还存在校准样片与测量样片分析层密度不一致导致的校正误差。为此,采用UniQuant软件中的扩展基本参数法,确定相应的谱线重叠校正系数Kij(待测元素i、干扰元素j)和取样量,以粉末压片制样-X 射线荧光光谱法测定二氧化锆材料中二氧化锆的含量,可为相关产品的质量监控提供技术参考。
1 试验部分
1.1 仪器与试剂
ARL 9900型X 射线荧光光谱仪;IRIS Advantage ER/S型电感耦合等离子体原子发射光谱仪;XCY-80型半自动压样机。
二氧化锆标准物质GBW 06602;铈元素标准溶液,1.0 g·L-1;钕元素标准溶液,1.0 g·L-1;硼酸为分析纯;三氧化钼纯度为99.99%;三氧化二铝样品纯度为99%。
1.2 仪器工作条件
X 射线荧光光谱仪仪器工作条件见表1,其中SC、FPC分别为闪烁计数器、流气正比计数器。

表1 X射线荧光光谱仪工作条件Tab.1 Working conditions of X-ray fluorescence spectrometer
1.3 试验方法
将样品研磨成粒径小于75 μm 的粉末,于105 ℃加热2 h。将2 g硼酸作为衬底置于压样机压腔中,铺平,称取不少于2.00 g样品粉末均匀覆盖在硼酸上,在压力410 k N、保压时间20 s的条件下,压制成外径为40 mm 的样片,按仪器工作条件测定。以UniQuant软件中谱线重叠系数Kij校正锆对钼、铈和钕的谱线重叠干扰,并用归一法校正取样量引起的分析层密度差异(采用二氧化锆标准物质GBW 06602 对二氧化锆、二氧化硅、三氧化二铝、三氧化二铁、氧化钙和氧化镁进行单点校正,其他元素按UniQuant的理论系数计算)和计算二氧化锆的含量。
2 结果与讨论
2.1 取样量的选择
锆元素为重元素,其X 射线穿透深度大,如ZrKα线在铁基中的穿透深度约为235μm[13]。根据压片机模具的直径(40 mm)和烧结二氧化锆样品的密度(6.10 g·cm-3),计算单位质量氧化锆粉末的样片厚度,结果为130.5μm·g-1,并以此计算样片厚度。取样量对样片厚度有直接影响,进而影响二氧化锆的测定,因此考察了不同取样量对归一化前后样片中成分测定结果的影响,结果见表2。
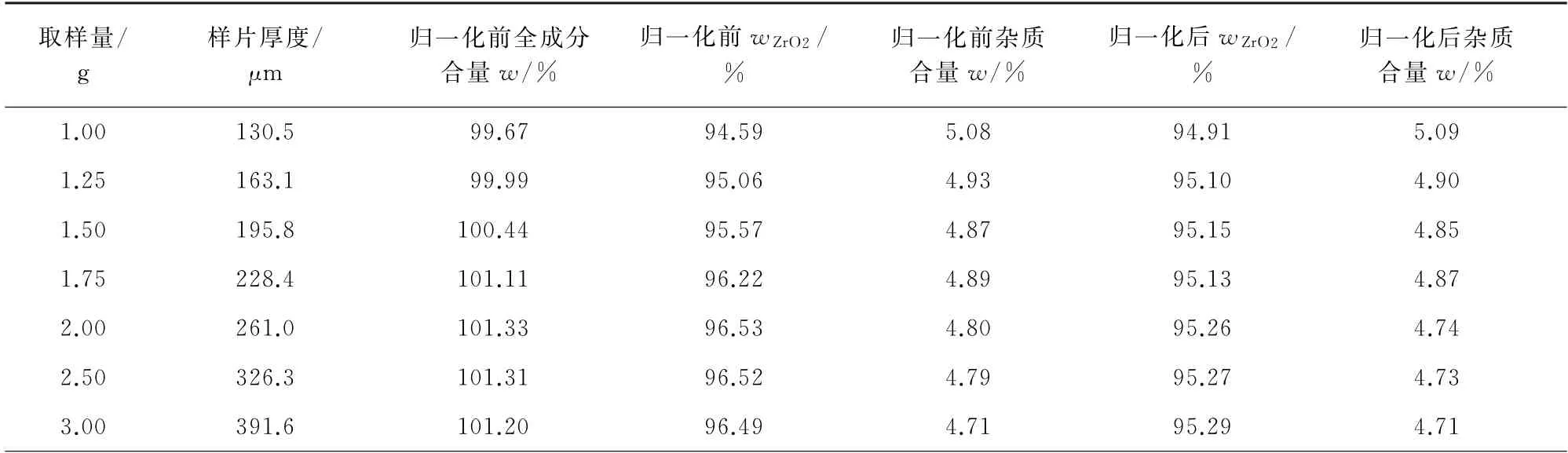
表2 取样量对测定结果的影响Tab.2 Effect of sample amount on the determined result

表2 (续)
以表2中取样量为横坐标,归一化前二氧化锆测定值为纵坐标作图,结果见图1。
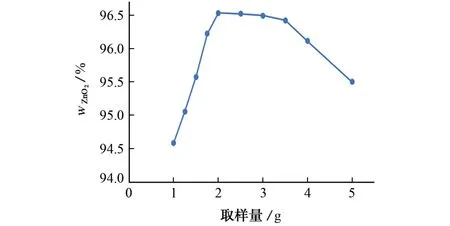
图1 取样量对归一化前二氧化锆含量的影响Fig.1 Effect of sample amount on the zirconium dioxide content before normalization
文献[14-15]认为:在不饱和厚度区,X 射线荧光强度随样品厚度的变化而变化;在饱和厚度区,X射线荧光强度不随样品厚度变化。使X 射线荧光强度不再随样品厚度增加而增加,并维持恒定的辐射穿透深度称为该辐射的临界厚度或无限厚。在常规X 射线荧光光谱法分析中,样片厚度都必须满足无限厚要求。表2 和图1 结果显示:取样量由1.00 g增至2.00 g时,样片归一化前全成分合量从99.67% 增至101.33%,二氧化锆质量分数由94.59%增至96.53%,合量随二氧化锆含量的增加而增加,说明全成分合量受二氧化锆含量影响较大;当取样量为1.00 g时,样片厚度显著低于锆的临界厚度(以X 射线入射角与辐射角均为45°计算,167μm),未达到常规分析要求。此外,由于样片很难被均匀压制,取样量较低的样片可能产生厚薄不均现象,而该现象可用逆光图显示,其中取样量为1.00,2.00 g时对应的图片见图2。
由图2可知:取样量为1.00 g时,样片中呈现出多个透光的浅色区域,浅色区域的样品厚度较小,产生锆的荧光产额较低,二氧化锆测定结果偏低;而取样量为2.00 g时,样片辐射面均匀度较好。因此,采用粉末压片法制样时,取样量应不小于2.00 g。

图2 取样量对样品辐射面均匀程度的影响Fig.2 Effect of sample amount on the radiant surface homogeneity
取样量为2.00~5.00 g时,样片厚度增加,锆元素的X 射线荧光强度随之降低,这和文献[14-15]中的规律不一致,推测随着取样量的增加,样片分析层密度逐渐降低,临界厚度内二氧化锆的含量逐渐降低,同时也不排除少数情况,即样品在压片过程中发生了剥离,致使二氧化锆测定结果偏低。由文献[16-18]可知,随着取样量的增加,样片的高径比增加,压制成型时受模壁摩擦力的影响,压力在轴向方向自上而下衰减,样片的分析密度逐渐减小,从而使X 射线激发的样品量减少,X 射线荧光强度降低。为了克服这种影响,文献[19]建议精确称量硼酸和样品粉末。然而,精确称量必然增加样片制备时间。即使精确称量,仍然可能存在卸压脱模过程中样片分析层剥离[20]等问题,使样片分析层密度减小,进而引起各成分的测定结果降低。
UniQuant是一款半定量分析软件,能够分析元素周期表中从氟到铀等78 种元素,并能同时测量64种纯物质的荧光强度。除此之外,该软件还建立了基于仪器灵敏度的数据库,涵盖1 500多条谱线的Kij。基于此功能,用标准物质校准后,即可选用合适的Kij进行定量分析;该软件还设计了归一法计算,无需精确称量即可消除由样片分析层密度不一致引起的误差,并能将X 射线荧光光谱仪未测项目(如碳、氢、氧、氮、硼、锂和灼烧减量)代入,参与归一法计算,使检测结果准确度提高。由表2结果可知:取样量为2.50~5.00 g时,归一法校正前后杂质合量变化不大,而二氧化锆测定值相差较大。计算归一法校正前后二氧化锆测定值的相对标准偏差(RSD),分别为0.44%,0.037%,说明归一法校正可显著提高主量成分分析的精密度,使取样量对二氧化锆测定的影响降低。因此,试验选择归一法校正检测数据。
2.2 锆对钼、钕和铈的谱线重叠干扰及干扰的校正
2.2.1 谱线重叠干扰
按照试验方法分析12个二氧化锆质量分数为50%~95%的样品,并采用二氧化锆标准物质GBW 06602对UniQuant软件中二氧化锆、二氧化硅、三氧化二铝、三氧化二铁、氧化钙和氧化镁进行单点校正。与差减法一样,UniQuant软件的归一法计算主量成分含量时的准确度受杂质含量测定误差和测定项目全面性的影响较大,因此试验在采用GBW 06602 进行单点校正后,以UniQuant软件的理论系数计算了其他成分的含量,各成分的结果列于表3,其中质量分数小于0.050%的杂质未列出。

表3 二氧化锆样品中主要成分的测定结果Tab.3 Determined results of main components in the zirconium dioxide samples
由表3可知,二氧化硅、三氧化二铝、三氧化二铁、氧化钙、氧化镁、二氧化钛和三氧化二钇含量变化范围较宽,而二氧化铪、三氧化钼、三氧化二钕和二氧化铈含量变化范围比较窄,均集中在某一测定值附近,且随二氧化锆含量的增加而增加。因此,铪、钼、钕和铈元素是来自样品还是由谱线重叠产生,需进一步试验。
为验证二氧化锆样品中是否含有铪、钼、钕和铈元素,参考文献[21]采用电感耦合等离子体原子发射光谱法分析样品。结果显示:二氧化铪的含量与X 射线荧光光谱法的接近,说明样品中含有铪元素,且HfLβ不受谱线重叠干扰;而三氧化钼、三氧化二钕和二氧化铈的质量分数均小于0.010%,和X 射线荧光光谱法的测定值相差较大,说明样品中钼、钕和铈元素含量很低,需进一步分析。
在UniQuant软件所得谱图中,钼、钕和铈元素的谱峰与锆元素的谱峰比较接近,而相应干扰在软件的谱线干扰库有列出,并在扩展基本参数法中给出了Kij。但由于标准样品数量较少及分析元素含量范围有限,UniQuant软件不能准确计算出相应的Kij。为此,需分析锆元素在钼、钕和铈元素测量谱线处的谱线重叠干扰,重新确定钼、钕和铈元素的Kij。为考察锆元素与MoKα线的重叠干扰,制备了三氧化钼质量分数均为0.5%的二氧化锆样品和三氧化二铝样品,并和二氧化锆样品、三氧化二铝样品和二氧化锆标准物质GBW 06602一起用试验方法分析,进行MoKα线扫描,得到的谱图列于图3(a)。为考察锆元素与CeLβ和NdLα线的重叠干扰,各滴加2滴钕元素标准溶液和铈元素标准溶液于三氧化二铝样片上,烘干后和二氧化锆样品一起按照仪器工作条件分析,分别进行CeLβ和NdLα线扫描,得到的谱图列于图3(b)、(c)和图3(d)、(e)。

图3 各样品经钼、钕和铈元素相应谱线扫描后的谱图Fig.3 Spectra of each sample after line scanning of corresponding spectral lines of Mo,Nd and Ce
由图3(a)可以看出:三氧化二铝样品的谱图在20.332°(MoKα的理论2θ)附近几乎为一条直线,而添加了0.5%三氧化钼的三氧化二铝样品在20.332°处可见明显发射峰;二氧化锆样品的谱图在20.332°附近(20.077°,ZrKβ的理论2θ)处出现1个强度很高的发射峰,因MoKα(20.332°)和ZrKβ(20.077°)相邻,推测锆含量较高时,产生了MoKα(20.332°)分析信号。由图3(b)、(c)和图3(d)、(e)可以看出:含铈和钕元素的三氧化二铝样品谱图和二氧化锆样品谱图在111.64°(CeLβ的理论2θ)附近和112.672°(NdLα的理论2θ)附近有发射峰,峰强度值都较小,均小于二氧化锆样品中对应2θ处的峰强度,表明二氧化锆样品中锆在铈和铌分析谱线处有重叠信号。
2.2.2 谱线重叠干扰校正
锆在钼、铈和铌分析谱线处的重叠信号会引起钼、铈和铌测定结果的偏高,进而使二氧化锆测定结果偏低。三氧化钼、二氧化铈和三氧化二钕含量可以通过UniQuant软件中的谱线重叠系数Kij来进行校正,而原始软件中的原Kij不准确,因此本工作以标准加入法先对Kij进行了校正,然后通过差减法重新计算12个二氧化锆样品(样品1~12)中二氧化锆的含量。结果显示,校正后重叠干扰物质三氧化钼、二氧化铈和三氧化二钕的质量分数均为0,其他杂质合量分别为3.40%,8.60%,7.88%,8.52%,6.67%,7.72%,45.44%,3.69%,9.23%,4.29%,8.68%,12.10%,二氧化锆质量分数分别为96.60%,91.40%,92.12%,91.48%,93.33%,92.28%,54.56%,96.31%,90.77%,95.71%,91.32%,87.90%。为了验证校正效果,对4个三氧化钼质量分数分别为0.1%,0.2%,0.3%,0.5%的样品9#进行分析,所得三氧化钼的质量分数为0.08%,0.19%,0.28%,0.52%,二氧化铈和三氧化二钕的质量分数均为0,其他杂质合量为9.29%,9.27%,9.12%,9.17%,二氧化锆的质量分数为90.63%,90.54%,90.60%,90.31%。从以上结果可以看出:校正后12个二氧化锆样品中二氧化锆质量分数和校正前的差值为0.71%,0.62%,0.63%,0.62%,0.64%,0.63%,0.20%,0.69%,0.61%,0.69%,0.61%,0.59%,和校正前三氧化钼、二氧化铈和三氧化二钕合量0.730%,0.688%,0.676%,0.686%,0.692%,0.706%%,0.417%,0.730%,0.672%,0.720%,0.693%,0.667%基本一致,且其他杂质合量在校正前后变化不大;添加三氧化钼的4个样品中三氧化钼的测定值和添加量基本一致,说明校正效果较好。
2.3 准确度和精密度试验
按照试验方法分析样品3,5,7,9和二氧化锆标准物质GBW 06602,每个样品制备5个平行样,计算测定值的RSD,并和熔融制样-X 射线荧光光谱法[22]结果比对。结果显示:本方法测定值的RSD分别为011%,0.15%,0.11%,0.15%,0.080%,说 明本方法的精密度较好;本方法二氧化锆的测定值分别 为92.19%,93.47%,54.55%,90.63%,99.42%,和熔融制样-X 射线荧光光谱法的结果(92.05%,93.28%,54.46%,90.47%,99.45%)基本一致,且两种方法所得的二氧化锆标准物质GBW 06602的测定值均在认定值的不确定度范围内,说明本方法的准确度较好。
本工作采用粉末压片法制样,通过UniQuant软件调整三氧化钼、二氧化铈和三氧化二钕的Kij,并用归一法消除样品分析层密度的影响,以X 射线荧光光谱法测定二氧化锆材料中二氧化锆的含量。方法检测的杂质种类较全面,且消除了干扰物质的谱线重叠干扰,准确度与精密度进一步提高,可为二氧化锆材料中二氧化锆含量的准确测定提供技术参考。
