磁性介孔Al2O3 负载Fe-N-C 催化剂的制备及其在卤代硝基苯转移加氢反应中的性能研究
2022-08-13李军旗陈朝轶兰苑培
周 丽, 李军旗*, 张 伟, 陈朝轶, 兰苑培
(1. 贵州大学 材料与冶金学院, 贵州 贵阳 550025;
2. 贵州省冶金工程与过程节能重点实验室, 贵州 贵阳 550025)
在催化剂制备中, 载体不但使催化剂具有合适的形状、 尺寸和机械强度, 还使活性组分分散在载体表面, 获得较高的比表面积, 从而提高催化剂的催化效率. 除此之外, 载体还可以阻止活性组分在使用过程中团聚, 提高催化剂的循环使用性[1-2]. 而氧化铝是一种多孔性高分散的固体材料, 具有稳定性高、 比表面积大、 成本效益低和制备工艺简单等特点, 因而被广泛用作催化剂的载体[3-5]. 活性氧化铝制备方法主要有拟薄水铝石脱水法和溶胶-凝胶法. 耿红娟[6]通过以氢氧化铝、 一水软铝石和拟薄水铝石为前驱体制备活性氧化铝球, 考察抗压强度和表面性能, 得出拟薄水铝石是制备高比表面积和大孔容活性氧化铝球的理想原料. Li等[7]通过溶胶-凝胶法和高温焙烧法自组装形成三维层级花状活性氧化铝, 保留了纳米级活性位点. 上述两种方法中前者经济实用, 生产出的氧化铝纯度高, 产品晶型好, 孔结构容易控制; 后者生产方法简单, 是目前工业领域的主流方法.
磁响应核和功能壳的磁性纳米复合材料具有独特的功能和可分离性[8-9]. Fe3O4具备强力磁活性和快速磁场响应等优异特性, 在引入磁场后易于回收,通常用作多相催化剂的载体或添加剂[10-12]. 介孔材料有特殊的比表面积和孔体积, 可控的孔径, 明确的有序介孔结构. 为了实现磁响应、 高孔体积和大的比表面积, 将介孔结构与磁性结合成磁介孔材料具有很高的应用价值[13-15]. Rezayati等[16]制备出Fe3O4@SiO2@GP/Picolylamine-Cu(II)催化剂用于芳香醛、 尿素和乙酰乙酸乙酯多种化合物的Biginelli反应, 表现出高转化率和强大的重复使用能力. Zhu等[17]制备了一种磁性Pt/NiO-Al2O3@Fe3O4催化剂在4-乙基酚、 4-甲基酚等酚类物质的加氢脱氧反应中表现出较高的活性. 因此, 磁性催化剂在吸附、 药物传递、 锂离子电池和催化等方面具有潜在的应用前景.
我们制备了以四氧化三铁为核, 氢氧化铝为壳的前驱体材料, 这种核壳型磁性介孔氢氧化铝微球具有较强的磁响应性、 定向可达介孔和高分散性.以三聚氰胺作为碳氮源, 过渡金属Fe为活性组分,通过焙烧得到氮掺杂碳的磁性介孔催化剂Fe-N-C/Fe3O4@nSiO2@Al2O3. 这种催化剂将磁芯封装在介孔壳内, 同时能够将Fe纳米粒子固定在孔内. 用该催化剂转移加氢卤代硝基苯时表现出优异的催化性能且没有发生脱卤现象, 并且催化剂能够多次循环使用. 因此, 本催化剂不仅具有纳米催化剂的优异特性, 还具有磁性分离特性, 在化学反应和分离过程中有很好的应用.
1 实验部分
1.1 实验原料
六水合氯化铁(FeCl3·6H2O), 乙二醇, 乙酸钠(NaAc), 硅酸四乙酯(TEOS), 铝酸钠(NaAlO2), 尿素(CO(NH)2), 四水合氯化亚铁(FeCl2·4H2O), 1,10-邻菲罗啉, 三聚氰胺及各种卤代硝基苯购自上海阿拉丁工业公司. 水合肼(N2H4·H2O)购自天津科密欧化学试剂有限公司. 其他所有试剂和溶剂均原样使用,未进一步纯化.
1.2 磁性介孔Fe3O4@nSiO2@Al(OH)3前驱体的制备
磁性Fe3O4的制备[18]: 取2.7 g FeCl3·6H2O溶于100 mL乙二醇中, 加入7.2 g NaAc剧烈搅拌, 转移到聚四氟乙烯反应釜中以200 ℃加热10 h后冷却至室温, 用去离子水和乙醇清洗后在真空干燥箱中60 ℃干燥12 h得到Fe3O4.
无孔Fe3O4@SiO2(记为Fe3O4@nSiO2)的制备: 取0.1 mg Fe3O4超声分散在50 mL盐酸溶液(2 mol/L)中,用去离子水彻底清洗后加入体积比为4∶1的乙醇和去离子水, 1 mL的浓氨水, 超声分散形成稳定的分散溶液, 再加入0.5 g TEOS搅拌6 h, 将得到的样品用去离子水和乙醇清洗后在真空干燥箱中以60 ℃干燥1 d得到Fe3O4@nSiO2.
Fe3O4@nSiO2@Al(OH)3前驱体的制备: 取0.2 g Fe3O4@SiO2超声分散在20 mL去离子水中, 1.0 g NaAlO2和2.9 g CO(NH)2溶解在30 mL去离子水中,将两个溶液混合, 剧烈搅拌呈均匀的悬浮液后转移到反应釜中以140 ℃加热12 h后分离, 用去离子水和乙醇清洗后在40 ℃的真空干燥箱中干燥12 h得到Fe3O4@nSiO2@Al(OH)3.
1.3 磁性介孔Fe-N-C/Fe3O4@nSiO2@Al2O3催化剂的制备
采用浸渍法制备Fe-N-C/Fe3O4@nSiO2@Al2O3催化剂: 取0.1 g Fe3O4@nSiO2@Al(OH)3超声分散在乙醇溶液中, 加入由FeCl2·4H2O和Phen配制得到的混合液Phen-Fe2+搅拌1 d(其中, Fe与Phen的摩尔比为1∶4, 且混合液中Fe2+的浓度为6 mg/mL, Phen-Fe2+与Fe3O4@nSiO2@Al(OH)3的重量比为Phen-Fe2+的理论负载量), 接着加0.1 g的三聚氰胺再搅拌1 d后旋转蒸发得到红黑色样品; 将样品在N2保护下焙烧2 h得到Fe-N-C/Fe3O4@nSiO2@Al2O3催化剂.
为了得到催化效果优良的催化剂, 我们对焙烧温度和Phen-Fe2+理论负载量进行了探索. 对应的催化剂命名为x-y-Fe-N-C/Fe3O4@nSiO2@Al2O3, 其中x为焙烧温度,y为Phen-Fe2+理论负载量.
1.4 卤代硝基苯的转移加氢
将5.0 mg的催化剂和0.5 mmol卤代硝基苯放置在一个反应管中, 加入5 mL乙醇和适量的N2H4·H2O超声分散均匀. 在80 ℃的油浴锅中加热并不断搅拌以开始转移加氢反应. 为了跟踪反应过程, 样品在一定时间内用注射器抽取, 用有机膜滤头过滤. 提取的样品用气相色谱分析. 每次反应后, 用乙醇洗涤催化剂, 干燥后再做循环实验, 测试催化剂的可重复使用性.
1.5 表征
利用扫描电子显微镜(SEM, ZEISS Sigma 300)和透射电子显微镜(TEM, Thermo Fisher)对样品进行了形貌表征. 在X PertPowder衍射仪上, 以Cu-Kα辐射为X射线源, 在2θ为5°~90°范围内进行了X射线粉末衍射(XRD)分析. 利用X射线光电子能谱(XPS, Thermo Scientific K-Alpha)分析了Fe-N-C/Fe3O4@nSiO2@Al2O3表面的电子态. 利用振动样品磁强计(VSM, LakeShore7404)测量Fe3O4、 Fe3O4@nSiO2、 Fe3O4@nSiO2@Al(OH)3和Fe-N-C/Fe3O4@nSiO2@Al2O3的磁性. 采用ASAP2460(micromertics)进行N2吸附-脱附等温线获得BET (Brunauer-Emmett-Teller, BET)比表面积和孔径分布. 采用气相色谱仪(GC-2014C, 色谱柱型号、 ID、 长度、 温度、内径、 膜厚分别是wondacap-5、 01、 30.00 m、 70 ℃、0.25 mm、 0.25 μm, 汽化室温度: 250 ℃, 检测室温度: 280 ℃)测定卤代硝基苯转移加氢反应效果.
2 结果讨论
2.1 Fe-N-C/Fe3O4@nSiO2@Al2O3的表征分析
图1 是Fe-N-C/Fe3O4@nSiO2@Al2O3催化剂的制备流程. 为了解样品的微观形貌, 对样品做了TEM和SEM, 如图2所示. 图2a是分散性非常高的磁性Fe3O4的TEM 图. 图2b 是Fe3O4@nSiO2的TEM 图,可以看出SiO2的厚度约为30 nm. 图2c是Fe3O4@nSiO2@Al(OH)3的TEM图像, 可以清楚地观察到磁铁矿芯夹层结构, 中间层为SiO2, 外层为海绵状Al-(OH)3. 图2d-f是Fe3O4@nSiO2@Al(OH)3前驱体不同放大倍数的SEM图, SEM图像显示Fe3O4@nSiO2@Al(OH)3微球大小和形状均匀, 而表面的Al(OH)3则是棒状. NaAlO2与尿素通过库仑力相互作用, 在Fe3O4@nSiO2微球表面协同组装为Al(OH)3形成有序的介孔结构. 在本合成体系中, 首先, 中间无孔SiO2可以避免磁铁矿在严苛环境下腐蚀. 其次, 介孔Al-(OH)3壳不仅提供了较高的比表面积, 而且能为纳米粒子的催化和封装提供了较大的可达孔容.
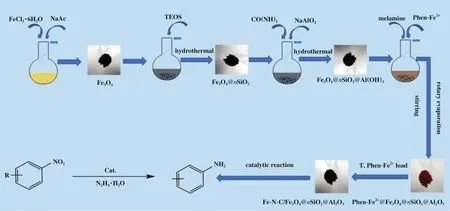
图1 Fe-N-C/Fe3O4@nSiO2@Al2O3催化剂的制备流程Fig.1 Preparation process of Fe-N-C/Fe3O4@nSiO2@Al2O3 catalyst

图2 (a) Fe3O4的TEM图; (b) Fe3O4@nSiO2的TEM图; (c) Fe3O4@nSiO2@Al(OH)3的TEM图;(d-f) Fe3O4@nSiO2@Al(OH)3不同放大倍数的SEM图Fig.2 (a) TEM image of Fe3O4; (b) TEM image for Fe3O4@nSiO2; (c) TEM image of Fe3O4@nSiO2@Al(OH)3;(d-f) SEM images of Fe3O4@nSiO2@Al(OH)3 at different magnifications
采用广角XRD分析了样品的物相. 图3a是Fe3O4、Fe3O4@nSiO2及Fe3O4@nSiO2@A(lOH)3的物相图, 由于Fe3O4的立方尖晶石结构, 所以在2θ=18.3°、30.1°、 35.5°、 35.1°、 43.2°、 47.2°、 53.5°、57.1°、62.7°处均可发现属于Fe3O4[18]的所有衍射峰, 分别对应Fe3O(4JCPDS No.88-0315)的(111)、(220)、( 311)、(222)、( 400)、( 422)、( 511)、( 440)和(533)晶面. 而Fe3O4@nSiO2和Fe3O4@nSiO2@A(lOH)3衍射峰与Fe3O4基本一致, 表明Fe3O4核与表面包裹的nSiO2和nSiO2@A(lOH)3结合良好, 说明成功合成了核壳结构的前驱体材料. 图3b是Phen-Fe2+理论负载量为6%时, 在不同温度下焙烧得到的样品的XRD图, 从图中可看出, 随着焙烧温度的升高, Fe3O4和Al2O(3JCPDS No.77-2135)衍射峰强度逐渐减小, 到700 ℃时消失; 当焙烧温度低于700 ℃时, 样品的衍射峰与Fe3N[19](JCPDS No.83-0879)几乎一致; 当焙烧温度为700 ℃及高于此温度时, 样品的衍射峰与Fe3C[20](JCPDS No.89-2722)一致. 图3c是焙烧温度为700 ℃时, Phen-Fe2+理论负载量不同时得到的样品的XRD图, 从图中可以看出当Phen-Fe2+理论负载量为6%时的衍射峰与Fe3C一致; 而Phen-Fe2+理论负载量为2%、 4%和8%时有Fe3O4、 Al2O3和Fe3N的衍射峰, 而Fe3O4的衍射峰相对Al2O3来说较弱, 原因是Fe3O4的衍射峰大部分被Al2O3的掩盖. 图3d是Fe-N-C/Fe3O4@nSiO2@Al2O3催化剂的能谱图, 对应的峰有C、 N、 O、 Si、 Al和Fe, 其成分组成与XRD中物相成分相对应.

图3 (a) Fe3O4, Fe3O4@nSiO2及Fe3O4@nSiO2@Al(OH)3的XRD图; (b) 不同焙烧温度的Fe-N-C/Fe3O4@nSiO2@Al2O3催化剂的XRD图;(c) Phen-Fe2+理论负载量不同时的XRD图; (d) 700 ℃-6%-Fe-N-C/Fe3O4@nSiO2@Al2O3催化剂的能谱图Fig.3 (a) XRD patterns of Fe3O4, Fe3O4@nSiO2 and Fe3O4@nSiO2@Al(OH)3; (b) XRD patterns of Fe3O4@nSiO2@Al(OH)3 catalysts at different calcination temperatures; (c) XRD patterns of different theoretical Phen-Fe2+ load;(d) the EDS spectrum of 700 ℃-6%-Fe-N-C/Fe3O4@nSiO2@Al2O3 catalyst
比表面积和孔结构是衡量催化剂具有良好催化效果的两个重要属性. 表1列出了样品的结构参数, 其中Fe3O4@nSiO2@Al(OH)3前驱体比表面积、孔体积、 平均孔径分别为45.7 m2·g-1、 0.2 cm3·g-1、14.9 nm. 而Fe-N-C/Fe3O4@nSiO2@Al2O3催化剂的比表面积、 孔体积、 平均孔径分别为129.7 m2·g-1、 0.3 cm3·g-1、 10.9 nm. 采用Brunauer-Emmett-Teller(BET)气体吸附法和Barrett-Joyner-Halenda(BJH)模型测量Fe3O4@nSiO2@Al(OH)3前驱体和Fe-N-C/Fe3O4@nSiO2@Al2O3催化剂的比表面积和孔径分布. 如图4a所示, 前驱体和催化剂的N2吸脱附等温线均为IV型等温线且存在滞后环, 表明样品中存在丰富的介孔结构. Fe-N-C/Fe3O4@nSiO2@Al2O3催化剂存在相对较强的滞后环是由于Phen-Fe2+与三聚氰胺吸附在前驱体上后在高温下焙烧还原, 该过程中三聚氰胺会产生腐蚀性气体NH3, 并从样品中释放出来, 形成更大的孔隙结构, 扩大比表面积, 导致催化剂中存在大量的介孔[21]. 样品吸附孔径分布如图4b可知,Fe3O4@nSiO2@Al(OH)3前驱体在1.7、 2.0、 2.7、 3.4和71.7 nm处有峰, 表明前驱体的孔径分布主要集中在较小的介孔范围内. Fe-N-C/Fe3O4@nSiO2@Al2O3催化剂在2.4和28.7 nm处有峰, 说明催化剂的介孔分布较集中, 从图中还可以看出催化剂的的孔径分布范围要比Fe3O4@nSiO2@Al(OH)3前驱体的广泛, 样品中除了有丰富的介孔结构, 还包含有少量的微孔和大孔, 这可能是由于氮掺杂碳层限制了活性组分Fe的团聚避免形成大尺寸的Fe.

表1 Fe3O4@nSiO2@Al(OH)3和700 ℃-6%-Fe-N-C/Fe3O4@nSiO2@Al2O3的结构参数Table 1 Structural parameters of Fe3O4@nSiO2@Al(OH)3 and 700 ℃-6%-Fe-N-C/Fe3O4@nSiO2@Al2O3

图4 Fe3O4@nSiO2@Al(OH)3和700 ℃-6%-Fe-N-C/Fe3O4@nSiO2@Al2O3吸脱附等温线图和孔径分布图Fig.4 Adsorption isotherm and pore size distribution of Fe3O4@nSiO2@Al(OH)3 and 700 ℃-6%-Fe-N-C/Fe3O4@nSiO2@Al2O3
用XPS研究了Fe-N-C/Fe3O4@nSiO2@Al2O3催化剂的元素组成、 化学状态和电子状态如图5. 从图5a的全谱图中可观察到C、 N、 O、 Al和Fe的峰. 图5b是C 1s的精细谱, 可拟合为284.8、 285.6和288.8 eV 3 个特征峰, 分别对应C=C/C-C、 C-N/C-O和C=O/Csp3-N键、 C-N键和Csp3-N的出现表明氮基体中掺杂了碳原子[19,22-24]. 图5c是是N 1s的精细谱, 可拟合成结合能分别为399.5、 399.5、 400.5、401.6和402.7 eV的5个峰, 分别对应吡啶N、 Fe-Nx、吡咯氮、 石墨氮和氧化氮, 结合图2b和图5e的分析, Fe-N-C/Fe3O4@nSiO2@Al2O3催化剂中的金属-Nx是Fe3N[25-28]. 图5d显示了与O 1s相关的电子结合能, 有3 个峰, 其中530.1 eV为Fe-O键, 531.4 eV为Al-O键, 533.2 eV为C-O-C键[19,23,28]. 图5e是Al 2p的精细谱, 结合能在75.4 eV处时对应Al2O3中Al的峰, 峰位没有左右移动, 说明Al的化学状态没有变化, 只有Al-O键[28]. 图5f是Fe 2p精细谱, 峰值710.3 eV是金属Fe或铁碳化物, 711.7 eV是Fe-Nx, 表明Phen-Fe2+与三聚氰胺在高温焙烧过程中被还原成Fe-N-C结构, 氮掺杂碳层将铁封装在前驱体孔道内[29-30]. 在结合能为710.9 和724.3 eV分别对应Fe2+2p3/2和Fe2+2p1/2. 713.8 和726.7 eV分别对应Fe3+2p3/2和Fe3+2p1/2, 表明Fe 2p1/2和Fe 2p3/2处的峰是Fe3O4. 而718.9和732.4 eV分别对应Fe2+和Fe3+的卫星峰.
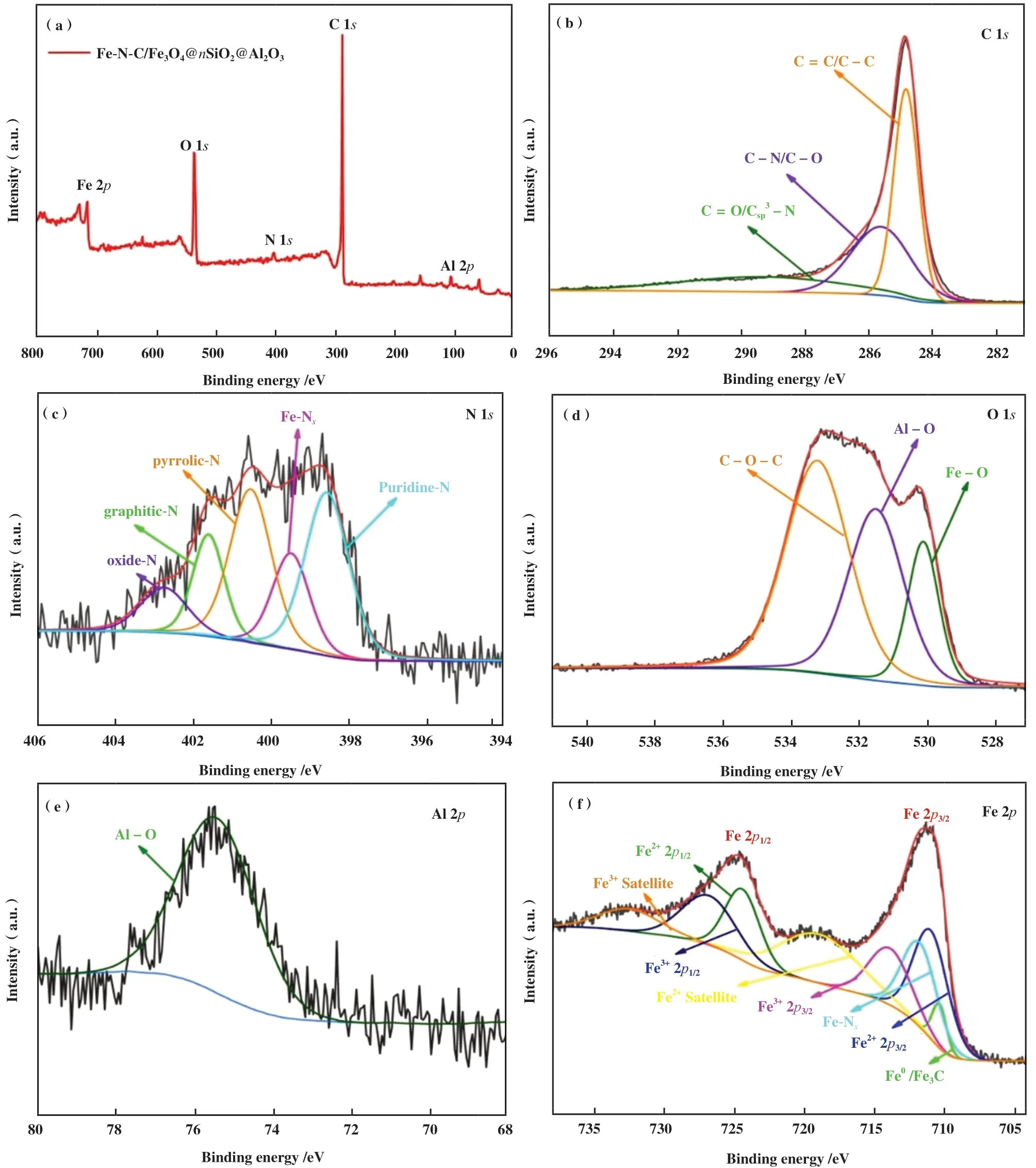
图5 (a) 700 ℃-6%-Fe-N-C/Fe3O4@nSiO2@Al2O3的XPS全谱图; (b) C 1s精细谱; (c) N 1s精细谱; (d) O 1s精细谱;(e) Al 2p精细谱; (f) Fe 2p精细谱Fig.5 (a) Wide-scan XPS spectrum of 700 ℃-6%-Fe-N-C/Fe3O4@nSiO2@Al2O3; (b) the C 1s; (c) the N 1s; (d) the O 1s;(e) the Al 2p; (f) the Fe 2p spectra
在常温下, 利用VSM分析Fe3O4、 Fe3O4@nSiO2、Fe3O4@nSiO2@Al(OH)3和Fe-N-C/Fe3O4@nSiO2@Al2-O3的磁性特征如图6 所示. 从图中可以看出, 所有的样品均表现出超顺磁性. 其中, Fe3O4@nSiO2(49.06 emu·g-1)的饱和磁化强度小于Fe3O4(83.05 emu·g-1), 这主要是由于SiO2包裹在Fe3O4颗粒上.Fe3O4@nSiO2@Al(OH)3(34.08 emu·g-1)的饱和磁化强度略小于Fe3O4@nSiO2, 这是由于Al(OH)3包裹在Fe3O4@nSiO2上. 而Fe-N-C/Fe3O4@nSiO2@Al2O3(62.75 emu·g-1)的饱和磁化强度高于Fe3O4@nSiO2@Al-(OH)3, 这是因为负载了Phen-Fe2+引入了Fe且有磁滞回线, 表明Fe-N-C/Fe3O4@nSiO2@Al2O3存在铁磁行为. 当施加外部磁场时, 溶液中均匀分散的样品迅速聚集在一边, 当磁铁被移开后, 摇动样品可使其分散. 这表明样品具有良好的磁响应性, 有助于磁性催化剂在实际应用过程中回收.
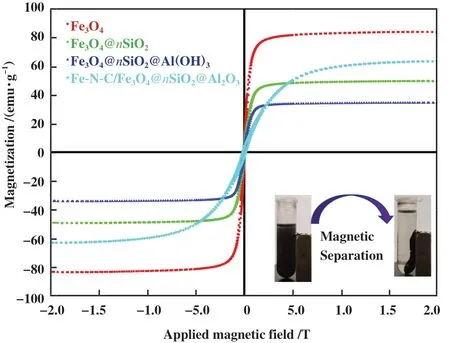
图6 Fe3O4、 Fe3O4@nSiO2、 Fe3O4@nSiO2@Al(OH)3和700℃-6%-Fe-N-C/Fe3O4@nSiO2@Al2O3的VSM曲线图(插图是外部磁铁的回收处理)Fig.6 The VSM curves of Fe3O4, Fe3O4@nSiO2, Fe3O4@nSiO2@Al(OH)3 and 700 ℃-6%-Fe-N-C/Fe3O4@nSiO2@Al2O3(illustration of the recycling of external magnets)
2.2 催化剂性能测试
对本研究制备的催化剂进行了催化性能测试, 以4-氯硝基苯(4-CNB)转移加氢为模型反应来探索最佳反应条件. 以乙醇为溶剂, 首先确定不同N2H4·H2O用量时催化剂的催化性能, 如表2(表2,Entry 1). 可得出无N2H4·H2O时催化反应几乎不发生, 表明该催化反应需要还原剂(表2, Entry 2, 3, 4,5). 表明随着N2H4·H2O的用量增加, 4-CNB的转化率越高. 当N2H4·H2O用量高于2 mmol时, 90 mim内4-氯苯胺(4-CPA)的产率均大于99.9%, 几乎完全反应. 综上, N2H4·H2O用量为2 mmol即可.

表2 700 ℃-6%-Fe-N-C/Fe3O4@nSiO2@Al2O3转移加氢4-CNB时N2H4·H2O用量筛选Table 2 N2H4·H2O dosages screening of 700 ℃-6%-Fe-N-C/Fe3O4@nSiO2@Al2O3 transfer hydrogenation of 4-CNB
表3是Phen-Fe2+理论负载量为6%时样品以不同温度焙烧的催化实验. 从表中可得出当焙烧温度为700 ℃时, 4-CPA的产率最高, 这缘于700 ℃时Phen-Fe2+和三聚氰胺被充分还原成Fe-N-C结构, 这给活性组分Fe提供了大量活性位点. 而当焙烧温度高于700 ℃时4-CPA的产率逐渐降低, 这是由于焙烧温度过高导致前驱体结构坍塌, 减少了活性组分Fe的附着位点. 我们还将前驱体Fe3O4@nSiO2@Al(OH)3以700 ℃焙烧成Fe3O4@nSiO2@Al2O3, 再负载Phen-Fe2+. 结果表明4-CPA的产率几乎减半, 原因是前驱体焙烧脱水后导致介孔和比表面积缩小,给Phen-Fe2+提供的活性位点少. 综上所述, 前驱体Fe3O4@nSiO2@Al(OH)3在负载活性组分时不用焙烧,且催化剂的焙烧温度选择700 ℃合适.

表3 Fe-N-C/Fe3O4@nSiO2@Al2O3转移加氢4-CNB时焙烧温度的筛选Table 3 Calcination temperature screening of Fe-N-C/Fe3O4@nSiO2@Al2O3 transfer hydrogenation of 4-CNB
表4是焙烧温度为700 ℃时, 样品的Phen-Fe2+理论负载量为2%、 4%、 6%和8%的催化实验. 当Phen-Fe2+理论负载量低于6%时4-CNP的产率较低,原因是活性组分Fe的量少不足以支撑催化反应完全进行; 当Phen-Fe2+理论负载量为8%时, 由于活性组分过多而团聚, 苯胺的产率也不高. 而当Phen-Fe2+理论负载量为6%时, 在丰富的介孔结构和大的比表面积存在的前提下, 活性组分Fe尺寸适宜且均匀分布在介孔结构内, 使得催化剂具有良好的催化效果. 因此, Phen-Fe2+理论负载量为6%最为合适.

表4 Fe-N-C/Fe3O4@nSiO2@Al2O3转移加氢4-CNB时Phen-Fe2+理论负载量的筛选Table 4 Phen-Fe2+ theoretical loading screening of Fe-N-C/Fe3O4@nSiO2@Al2O3 transfer hydrogenation of 4-CNB
表5是催化剂催化卤代硝基苯的研究结果. 从表5可以得出, 该催化剂在卤代硝基苯的转移加氢制卤代苯胺的反应中表现出优异的活性和选择性,反应过程没有发生明显脱卤的现象.

表5 700 ℃-6%-Fe-N-C/Fe3O4@nSiO2@Al2O3催化剂对各种卤代硝基苯的转移加氢Table 5 The transfer hydrogenation of various halogenated nitrobenzenes catalyzed by
我们取2.5、 5.0、 7.5和10 mg的催化剂转移加氢4-CNB来研究反应的动力学. 为了消除N2H4·H2O的影响, 将N2H4·H2O的用量扩大5 倍, 4-CNB的转化率和4-CPA的产率随反应时间变化如图7a-d所示.结果表明, 催化剂用量高于5.0 mg时, 4-CNB在60 min内可以完全转化, 说明催化剂用量越大反应速率越快, 催化剂的选择性升高, 原因是催化剂用量越大, 可提供更多的活性位点. 由于N2H4·H2O的用量远大于4-CNB, 所以反应体系中N2H4·H2O的浓度可认为是不变的. 因此, 可以用准一级动力学方程来评价该反应的动力学过程. 表观速率常数k的计算公式为: ln(Ct/C0)=k·t, 其中C0为4-CNB的初始浓度, Ct为反应时间t (min)时4-CNB的浓度. 如图7e计算了催化剂用量为2.5、 5.0、 7.5和10 mg时的线性斜率分别为0.0182、 0.1481、 0.1652和0.2292 min-1,展现出一个较好的反应速率, 且随着催化剂用量的增加反应速率越快, 其中5 mg的反应速率约为2.5 mg的9倍. R2则分别为0.9880、 0.9643、 0.9457和0.9891,表明具有一个相对较高的相关性.
图7 在不同的700 ℃-6%-Fe-N-C/Fe3O4@nSiO2@Al2O3用量下, 4-CNB含量和4-CPA产率随时间的变化曲线(a) 2.5 mg; (b) 5.0 mg; (c) 7.5 mg; (d) 10.0 mg; (e) 不同用量的700 ℃-6%-Fe-N-C/Fe3O4@nSiO2@Al2O3转移加氢4-CNB动力学研究Fig.7 Conversion of 4-CNB and 4-CPA yield versus time at different dosages of 700 ℃-6%-Fe-N-C/Fe3O4@nSiO2@Al2O3(a) 2.5 mg; (b) 5.0 mg; (c) 7.5 mg; (d) 10.0 mg; (e) kinetics of 4-CNB transfer hydrogenation of 700 ℃-6%-Fe-N-C/Fe3O4@nSiO2@Al2O3 Reaction conditions: 5 mg catalyst, 0.5 mmol substrate, 10 mmol N2H4·H2O and 5 mL ethanol, reaction temperature 80 ℃
2.3 循环使用性测试
催化剂的稳定性是评价催化剂性能的重要因素, 因此, 我们使用催化剂转移加氢4-CNB进行了循环反应的研究. 在每次反应结束后, 利用外加磁场从反应溶液中回收催化剂, 用无水乙醇洗涤数次后, 放入真空干燥箱中常温干燥, 以进行下一次的循环实验. 如表6所示, 在连续7次循环测试后,4-CPA的产率仍高于85%, 且选择性高于99.9%. 因此, 本催化剂表现出较好的稳定性.

表6 700 ℃-6%-Fe-N-C/Fe3O4@nSiO2@Al2O3对4-CNB转移加氢的可重复使用性Table 6 The reusability of the 700 ℃-6%-Fe-N-C/Fe3O4@nSiO2@Al2O3 catalyst for the transfer hydrogenation of 4-CNB
3 结论
本研究制备了磁性介孔Fe3O4@nSiO2@Al(OH)3前驱体, 通过在其孔道内负载Phen-Fe2+制备了Fe-N-C/Fe3O4@nSiO2@Al2O3磁性介孔催化剂. 该催化剂在焙烧温度为700 ℃, Phen-Fe2+理论负载量为6%时,对卤代硝基苯的转移加氢表现出良好的催化活性.其中, Phen-Fe2+配体和三聚氰胺在高温焙烧后形成Fe-N-C结构可以将Fe活性物种稳固在前驱体的孔道内, 避免因反应过程中活性物种的脱落而降低催化剂的活性, 从而提高催化剂的稳定性. 该催化剂还具有很强的磁响应, 易于回收, 且对卤代硝基苯的转移加氢有很高的活性和选择性, 可重复使用至少7次. 本研究制备的磁性介孔铁基催化剂价廉、环保, 在磁性介孔纳米技术方向有很大的应用潜力.
