基于生物信息学分析FAM110A在乳腺癌中的表达及临床意义
2022-08-12马小雯杨巍郝靓
马小雯,杨巍,郝靓
(1.中国医科大学法医学院化学教研室,沈阳 110122;2.辽宁省法医学生物证据重点实验室,沈阳 110122;3.中国医科大学司法鉴定中心,沈阳 110122;4.中国医科大学临床二系,沈阳 110122;5.辽宁省健康产业集团本钢总医院感染部,辽宁 本溪 117000)
乳腺癌是全球发病率最高的癌症。2020年公布的全球癌症数据显示,乳腺癌新发病例高达226万例[1]。乳腺癌的早期症状多不明显,进展迅速,晚期可发生远端转移,直接威胁患者生命。浸润型乳腺癌是乳腺肿瘤中高发的类型,其分化程度低,预后差,以浸润性导管癌最为常见[2]。目前,虽然乳腺癌得到了广泛研究,但发病机制仍不明确。
FAM110A定位于20号染色体的短臂。2007年,HAUGE在运用酵母双杂交方法筛选参与细胞周期进程的中心体、微管相关蛋白的实验中首次发现了FAM110家族蛋白。FAM110家族有3个成员定位于中心体,协助细胞周期的进展并参与微管的组织[3]。FAM110A的表达受到细胞周期的调控,与肝癌和前列腺癌的发生发展相关[4-5]。目前FAM110A在乳腺癌中鲜有研究,其作用机制尚不明确。本研究利用多种数据库进行数据挖掘,分析FAM110A在乳腺癌中的表达及对预后的影响,为进一步研究FAM110A在乳腺癌发生发展中的作用机制和治疗提供线索和依据。
1 材料与方法
1.1 数据来源
1.1.1 Oncomine数据库的数据提取和分析:在Oncomine数据库(https://www.oncomine.org/resource/login.html)中根据本研究的需求设定筛选条件。(1)Gene:FAM110A;(2)Analysis type:cancer vs normal analysis;(3)Cancer type:breast cancer;(4)Data type:All;(5)P< 0.01,fold change >2,gene rank=top10%。
1.1.2 GEPIA数据库的数据提取及患者预后分析:利用GEPIA数据库(http://gepia.cancer-pku.cn/detail.php)对FAM110A的表达与分期和预后的关系进行预测。设置分期筛选条件为(1)Gene:FAM110A;(2)Datasets selection:BRCA;(3)Log Scale:yes。设置预后筛选条件为(1)Gene:FAM110A;(2)Methods:Overall Survival;(3)Group cutoff:median;(4)Hazards ratio:yes;(5)95% confidence interval:yes;(6)Axis units:months。
1.1.3 UALCAN数据库的数据提取和分析:利用UALCAN数据库(http://ualcan.path.uab.edu/)补充验证FAM110A在肿瘤不同分期、不同分子分型中的表达差异,并探究FAM110A表达与基因甲基化水平的关系。设置筛选条件为(1)Enter gene symbol:FAM110A;(2)TCGA dataset:Breast invasive carcinoma;(3)Links for analysis:Expression;(4)based on:individual cancer stages/major subclasses。
1.1.4 标本来源:利用人类蛋白图谱(human protein atlas,HPA)数据库(https://www.proteinatlas.org/)获取FAM110A在乳腺癌组织和正常组织中的免疫组织化学染色图片。
1.1.5 蛋白-蛋白相互作用网络分析:通过STRING数据库(https://www.string-db.org/)预测FAM110A相互作用蛋白。
1.1.6 共表达基因聚类分析及分子调控网络构建:从cBioPortal平台(http://www.cbioportal.org/)获 取TCGA数据库中FAM110A在乳腺癌中的共表达基因,然后运用DAVID数据库(https://david.ncifcrf.gov/)对相关度较高的基因进行基因本体论(gene ontology,GO)及京都基因和基因组(Kyoto encyclopedia of gene and genomes,KEGG)富集分析。采用STRING数据库构建共表达分子调控网络,利用Cytoscape中的Cytohubba插件计算共表达分子网络中节点连接度,选取连接度最高的10个节点。
1.2 统计学分析
采用GraphPad Prism 9软件行统计学分析,采用t检验统计乳腺癌与正常组织之间FAM110A的表达差异。FAM110A不同病理分期间的表达差异比较采用单因素方差分析。采用Kaplan-Meier法进行生存分析,组间生存率采用log-rank检验,P< 0.05为差异有统计学意义。
2 结果
2.1 FAM110A在所有肿瘤中的表达
根据设置的筛选条件,Oncomine数据库共有205项涉及FAM110A在不同肿瘤与正常组织比较的研究结果。其中10项研究有统计学意义。FAM110A在所有肿瘤中8项呈高表达,2项低表达。在乳腺癌中6项呈高表达。在GEPIA数据库中FAM110A在11种肿瘤组织中呈显著高表达。见图1。
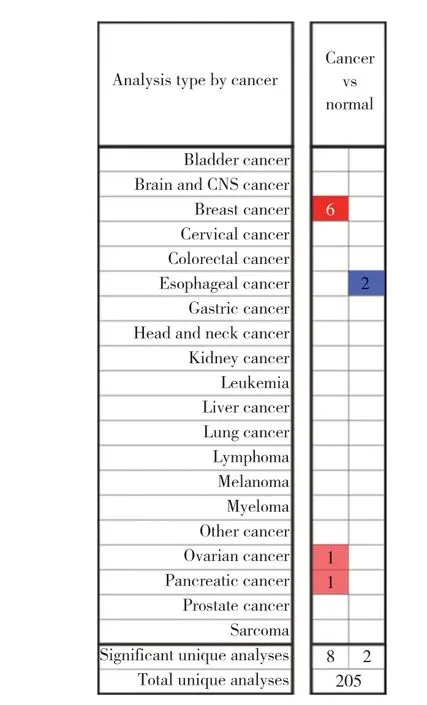
图1 FAM110A在所有肿瘤中的表达Fig.1 Expression of FAM110A in all cancer types
2.2 FAM110A在乳腺癌中高表达
按照筛选条件检索GEPIA数据库,结果显示,乳腺癌组织与正常组织相比,其FAM110A表达显著上调,差异有统计学意义(P< 0.05),见图2A。
检索HPA数据库,共得到3例FAM110A正常乳腺组织样本中的免疫组织化学图片,12例乳腺癌组织样本中的免疫组织化学图片(图2B)。乳腺癌样本中FAM110A呈高表达。在细胞内,FAM110A高表达于G2、S期,表达水平与细胞周期密切相关(图2C)。
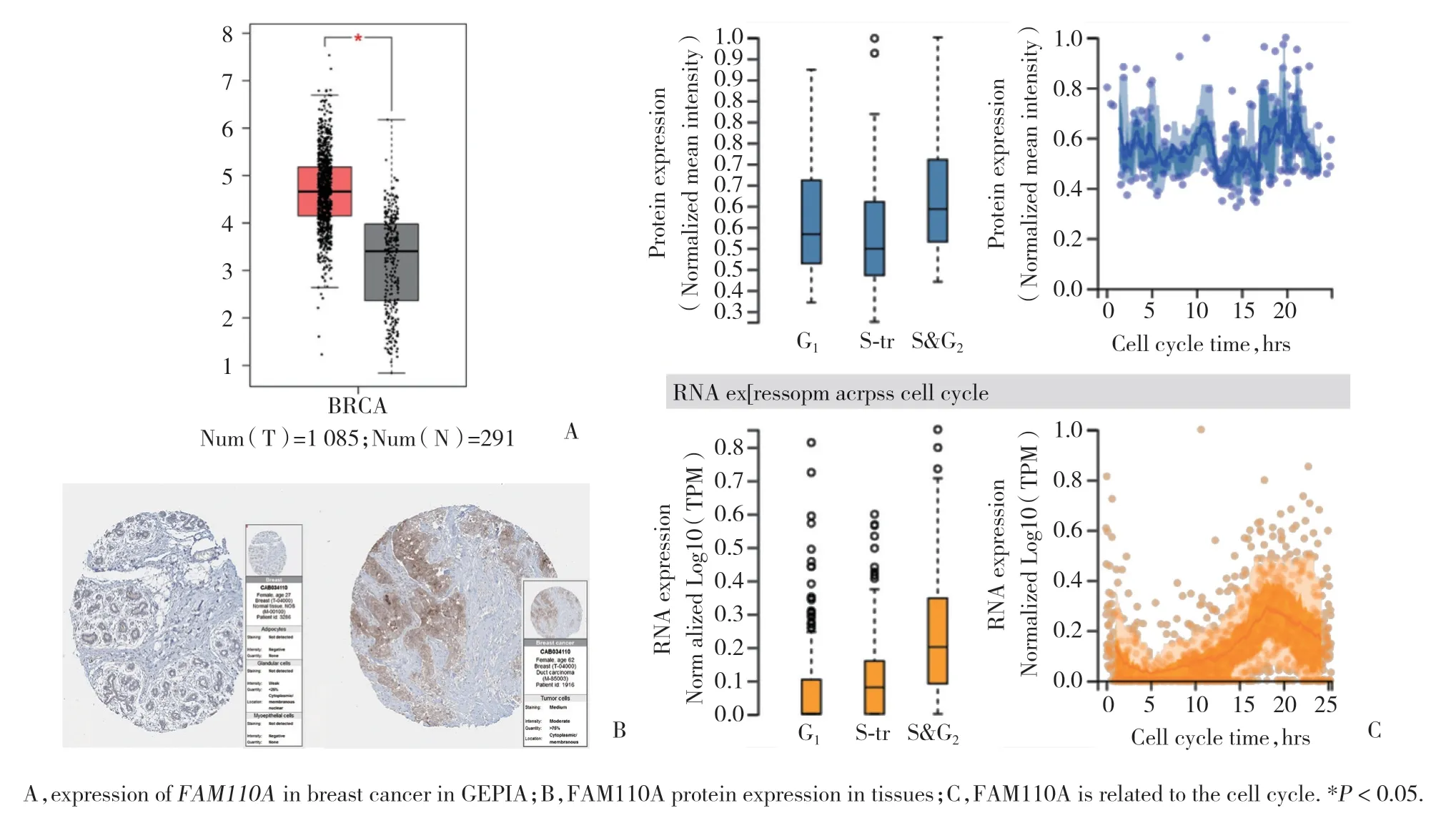
图2 FAM110A在乳腺癌中高表达Fig.2 FAM110A is highly expressed in breast cancer
2.3 FAM110A在不同分子分型乳腺癌中的表达情况
利用UALCAN数据库针对乳腺癌的分子分型进行在线分析。结果显示,与正常组织相比,FAM110A在Luminal、HER(2+)、三阴性乳腺癌中的表达量均呈高表达(P均<0.05)。且除去Luminal与HER(2+)之外,各个分型之间FAM110A表达量差异均有统计学意义,见图3。
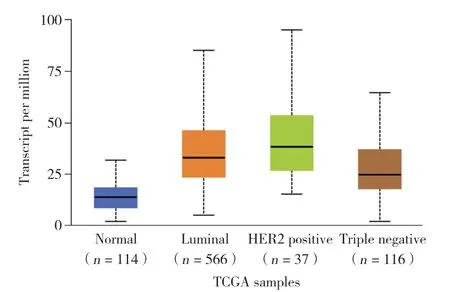
图3 FAM110A在不同分型乳腺癌中表达水平Fig.3 Expression of FAM110A in different types of breast cancer
2.4 FAM110A的表达与预后的相关性
运用GEPIA分析FAM110A表达水平与乳腺癌患者预后的关系,结果显示,FAM110A高表达组总生存期(overall survival,OS)劣于低表达组(HR=1.4,P=0.043),见图4A。在GEPIA数据库中在线分析FAM110A表达与乳腺癌分期的关系,结果显示,不同分期之间FAM110A的表达差异有统计学意义(F=2.84,P=0.023 3),见图4B。利用UALCAN数据库在线分析得到相似结果。
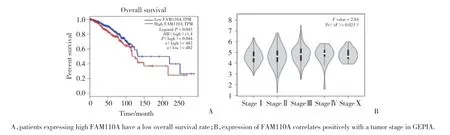
图4 FAM110A高表达提示不良预后Fig.4 High level of FAM110A expression is associated with poor prognosis
2.5 FAM110A共表达网络的构建及功能分析
运用STRING数据库得到FAM110A蛋白-蛋白相互作用网络图,富集P值为0.028 9,与FAM110A存在相互作用的蛋白节点有10个,分别为FAM19A1、SMIM13、CSPP1、GPANK1、IL9、ANKRD9、ZDHHC18、TMEM132D、FAM135B和KCTD3(图5)。检索cBioportal数据库,乳腺癌中FAM110A的共表达基因共有20 033个(P均<0.05)。相关度最高的10个基因为MRPS26、PPDPF、SNRPB、NSFL1C、SLC2A4RG、SCAND1、ITPA、MRPL41、ARRDC1、RBCK1。GO富集分析结果显示,FAM110A的共表达基因主要参与DNA模板转录调控,线粒体平移延伸等生物途径。KEGG通路富集分析结果显示,FAM110A的共表达基因主要参与泛素介导的蛋白质水解,癌症的中心碳代谢等通路。

图5 FAM110A共表达分子网络Fig.5 Co-expressed network of FAM110A
3 讨论
乳腺癌是起源于乳腺上皮组织的恶性肿瘤,发病率居全球女性恶性肿瘤的首位。其预后较好,患者生存率相对较高[6]。目前,乳腺癌主要根据临床病理和分子分型进行治疗,但三阴性乳腺癌和难治性多发耐药乳腺癌仍缺乏有效的治疗靶点[7]。因此,寻找乳腺癌的新治疗靶点和预后标志物对于乳腺癌的临床治疗极为重要。
FAM110A是FAM110家族成员,作为中心体相关蛋白,参与细胞周期调控[3]。有研究[4]表明,FAM110A的高表达与肝癌的发生和较差预后相关,但其在乳腺癌中的研究较少。本研究利用多种数据库分析FAM110A在乳腺癌中的表达水平及意义。
对Oncomine数据库中16项对比FAM110A在乳腺癌和正常组织中表达差异的研究进行分析,结果显示,FAM110A在乳腺肿瘤中呈高表达,可能诱导乳腺肿瘤的发生。FAM110A在不同分型乳腺癌中表达有统计学差异,可作为乳腺癌分型的标志物。分析HPA数据库中FAM110A的免疫组化结果,乳腺癌中FAM110A的表达高,定位于核质、胞质及囊泡中,高表达于S期和G2期。这与HAUGE等[3]研究结果一致。
GEPIA数据库分析结果显示,FAM110A随着乳腺癌分期的进展表达量显著上升,高表达的患者预后更差。甲基化是DNA的一种表观遗传修饰,对基因表达有影响。乳腺癌组织中FAM110A基因甲基化水平显著降低,提示甲基化修饰可能是上调FAM110A在乳腺癌中表达的重要因素。
STRING数据库分析结果显示,FAM110A可能与ANKRD9、FAM135B、CSPP1等分子存在相互作用。ANKRD9是一种E3泛素连接酶,通过蛋白酶体途径发挥抑癌作用[8]。FAM135B是一种细胞周期调控蛋白,可以正调控下游PI3K/Akt/mTOR通路[9]。有丝分裂相关蛋白CSPP1通过对于动点微管动态性的调节介导染色体的运动,保证有丝分裂的准确性[10]。FAM110A可与上述蛋白互作影响肿瘤发生。随后构建乳腺癌中FAM110A的共表达基因调控网络,通过研究关键节点的功能推断共表达基因网络参与的重要生物学途径。其中MRPL14作为连接度最高的节点,已被证实与乳腺癌相关[11-12]。
本研究对于FAM110A的共表达基因进行功能分析,探究其参与的生物途径及通路。GO富集分析结果显示,FAM110A的共表达基因主要参与DNA模板转录调控等生物途径。KEGG通路富集分析结果显示,FAM110A的共表达基因主要参与泛素介导的蛋白水解等通路。
综上所述,FAM110A在乳腺癌中呈高表达,且在不同分型乳腺癌中有统计学差异。FAM110A的表达水平与患者预后呈负相关,与分期呈正相关,可作为新的乳腺癌预后标志物。目前对于FAM110A在乳腺癌中作用机制的研究仍较少。本研究初步预测了FAM110A参与的生物途径及信号通路,为乳腺癌的诊治及机制研究提供了思路。
