儿童临床诊断不明呼吸系统疾病97 例基因二代测序结果分析
2022-08-09王峡代继宏田代印应林燕符州李莹
王峡 代继宏 田代印 应林燕 符州 李莹
重庆医科大学附属儿童医院呼吸科 国家儿童健康与疾病临床医学研究中心 儿童发育疾病研究教育部重点实验室 儿科学重庆市重点实验室(重庆 400014)
单基因病又称孟德尔遗传病,是由一对等位基因控制的疾病。呼吸系统单基因病包括直接起源于肺部(累及气道、肺实质和肺间质)的疾病和累及呼吸系统的其他遗传病[1]。近年来随着分子生物学技术的高速发展,靶向多基因面板测序和全外显子组测序(whole exome sequencing,WES)等二代测序技术已被广泛应用于儿童遗传病的诊断。随着越来越多的疑难罕见病在分子层面得以确诊,目前认为既往有低估其发病率的可能。但是与神经、肿瘤、内分泌、免疫等学科相比,基因二代测序(next generation sequencing,NGS)在辅助诊断儿童呼吸系统疾病中的应用尚处于起步阶段,其作用需要进一步探索。本研究对97例临床诊断不明的呼吸系统疾病患儿的临床表现及基因二代测序结果进行回顾性分析,旨在研究NGS在儿童呼吸系统疑难疾病中的诊断作用。其中测序结果根据美国医学遗传学与基因组学学会(American College of Medical Genetics,ACMG)对基因变异解读指南,利用多种生物信息学软件结合临床表型分析数据,从而明确致病/可能致病性基因变异。
1 对象与方法
1.1 研究对象
回顾性收集2016年1月—2021年10月年重庆医科大学附属儿童医院呼吸中心收治的以呼吸系统表现为主的患儿的临床资料。纳入标准:①年龄0~18岁;②临床诊断不明或常规治疗效果欠佳;③符合基因检测指征并完善了基因检测。排除标准:病例资料有缺失者。
基因检测指征:①反复呼吸道感染/慢性咳嗽常规抗感染治疗效果不佳,伴/不伴内脏转位,临床疑诊原发性纤毛运动障碍(primary ciliary dyskinesia,PCD)、囊性纤维化(cystic fibrosis,CF);②不明原因气促、呼吸窘迫,胸部影像学改变为弥漫性肺间质病变;③不明原因支气管扩张;④起病年龄小,肺炎重且合并其他系统受累(如发育迟缓、特殊面容)。
本研究获医院医学伦理委员会批准[批准号:(2022)年伦审(研)第(6)号],同时患儿监护人签署知情同意书。
1.2 方法
1.2.1 临床资料收集 使用标准化表格收集数据,包括一般人口学资料、临床表现、实验室检查结果、影像学检查结果、基因检测结果等。数据从临床记录中进行回顾性检索,或通过联系患儿家属获得。
1.2.2 基因检测方法 抽取患儿外周血2 mL,乙二胺四乙酸抗凝,行WES或呼吸系统疾病基因包测序(带或不带拷贝数变异分析),通过 Sanger测序进行验证并分析变异来源。基因检测阳性结果的判定标准:按照ACMG标准,变异符合分离规律,即在显性遗传模式的基因中检测到杂合变异,或在隐性遗传模式的基因中检测到纯合子/复合杂合变异。
1.3 统计学分析
采用SPSS 26.0 统计软件进行数据分析。非正态计量资料以中位数(M)(全距)表示,计数资料以例数(百分比)表示。
2 结果
2.1 一般临床资料
共102例患儿符合纳入标准,排除5例临床资料不全者,最终纳入97例患儿进行基因二代测序。男57例、女40例,样本采集时中位年龄为1.9(7.6)岁。80例进行了WES检查,17例行呼吸系统疾病基因包检查,9例行拷贝数变异分析,65例患儿父母同时接受基因测序。根据患儿临床表型及影像学特点可初步分为肺实质病变和肺间质病变两组。其中89例表现为慢性湿性咳嗽,CT提示支气管扩张、肺不张、肺实变,纳入肺实质病变组;8例表现为进行性加重的气促、发绀,不伴明显咳嗽,CT 提示广泛肺间质改变,纳入肺间质病变组。
2.2 临床特征
通过基因检测确诊呼吸系统单基因病共31例,诊断率32.0%,男16例、女15例,中位年龄4.0(7.6)岁。其中肺实质病变组28例,包括PCD 20例,CF 4例,原发性免疫缺陷病(primary immunodeficiency disease,PID)2例,神经肌肉疾病(neuromuscular disease,NMD)2例;肺间质病变组3例(均为肺表面活性物质代谢障碍)。2018年之前诊断4例,2018年之后诊断27 例。PCD、CF 患儿均以反复咳嗽、咳痰为主要表现,部分PCD 患儿合并有鼻窦炎、中耳炎,肺表面活性物质代谢障碍患儿以气促、发绀为主要表现,PID患儿以反复呼吸道感染为主要表现,无明显其他系统感染病史及阳性家族史,NMD患儿以反复呼吸道感染伴/不伴肌无力为主要表现。经胸部CT 证实,PCD、CF 患儿主要累及肺实质,肺表面活性物质代谢障碍则以肺间质受累为主。NGS 检测结果阴性患儿以反复呼吸道感染和慢性咳嗽表现为主,伴/不伴支气管扩张;部分患儿表现为不明原因气促、呼吸窘迫,伴/不伴肺部广泛间质改变。见表1、表2。典型病例的影像学表现见图1。

图1 典型病例胸部CT 表现
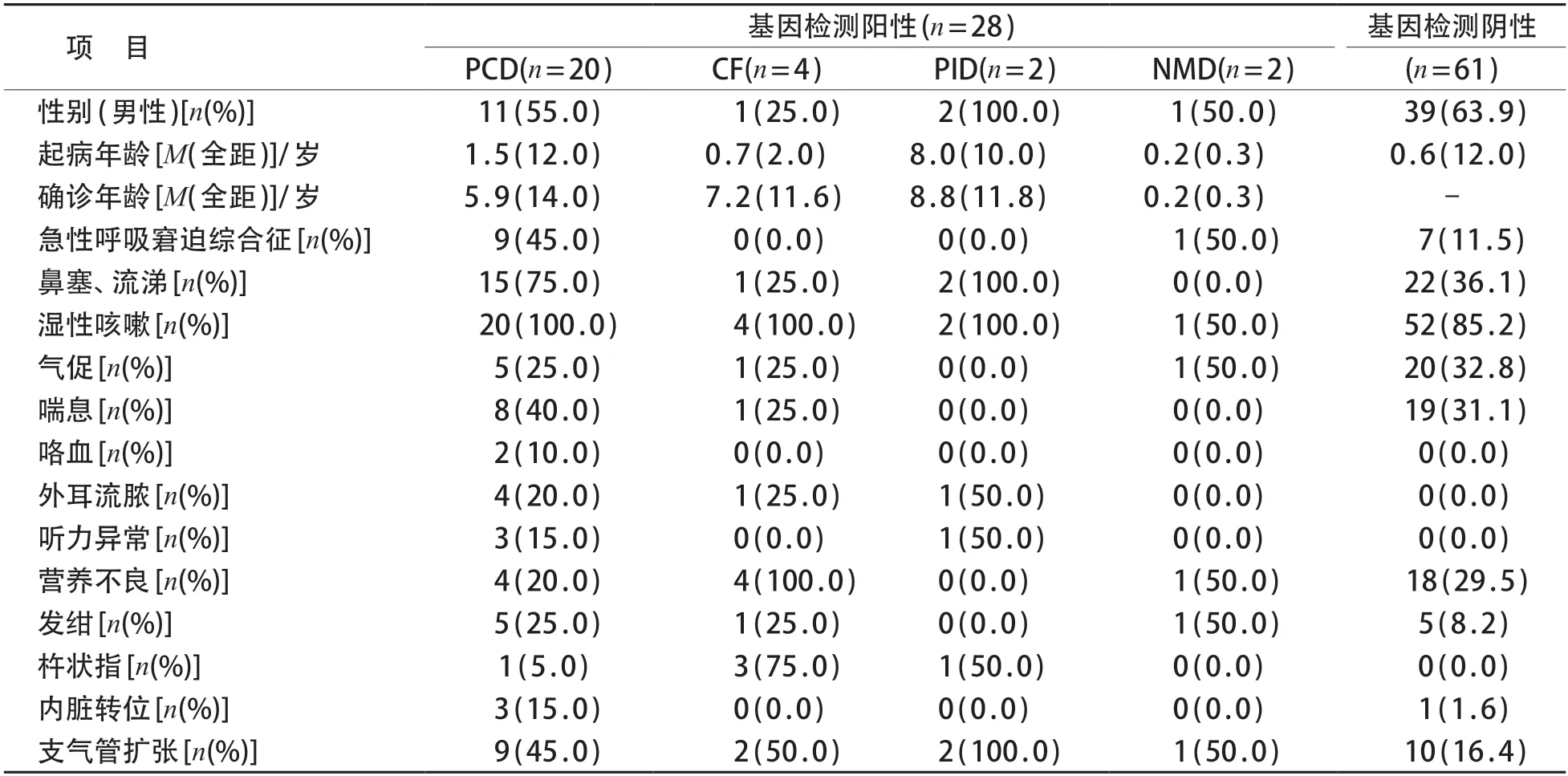
表1 肺实质病变组患儿临床特征

表2 肺间质病变组患儿临床特征
2.3 基因检测结果
PCD 主要为常染色体隐性遗传,18 例为复合杂合变异(HYDIN7例、CCNO3例、CCDC402例、DNAH12例、DNAAF31例、DNAI21例、DNAH111例、RSPH4A1例);1例为DNAI2基因纯合变异;1 例存在X 连锁PIH 1 D 3基因半合子缺失。以错义变异为主,少部分为移码变异、剪接变异、无义变异。肺表面活性物质代谢障碍均为SFPTC基因杂合变异,均为常染色体显性遗传。CF 4 例均为常染色体隐性遗传,3 例为CFTR基因复合杂合变异,1例为CFTR基因c.4056 G>C 纯合变异。2 例PID 患儿均为常染色体显性遗传,分别为PIK 3 CD基因杂合变异所致PI 3 Kδ 过度活化综合征(activatedphosphoinositide 3-kinase-δ syndrome,APDS)、CXCR4基因杂合变异所致WHIM综合征。2例NMD患儿分别为MTM1基因变异所致X连锁隐性遗传致中央核肌病、SMN 1基因纯合缺失致常染色体隐性遗传进行性脊肌萎缩症。
确诊的31例患儿(包括2例纯合子),父母均非近亲婚配,1例患儿 Xq22.3 区域存在半合子缺失,其弟弟同样检测出该缺失(因未在本院就诊缺乏临床数据),进一步分析该变异来自其母亲。另有2 例患儿为同胞姐弟,临床表型相似,均起病于新生儿期,合并有鼻窦炎、中耳炎,肺功能均提示轻度阻塞,但弟弟有卵圆孔未闭、全内脏转位,基因检测均提示CCDC 40复合杂合变异。其余患儿父母、家庭其他成员均无类似病史。
2.4 相关实验室检查
31例患儿均完善了痰液或支气管肺泡灌洗液的病原学培养,20例呈阳性(64.5%),最常见的细菌分离株是流感嗜血杆菌(9例)和肺炎链球菌(5例)。9例PCD患儿肺功能检查提示为阻塞性通气功能障碍,2 例肺表面活性物质代谢障碍患儿肺功能检查提示限制性通气功能障碍。2例PID患儿进行了免疫功能筛查(免疫球蛋白、白细胞吞噬功能检测、淋巴细胞分类及计数),免疫球蛋白水平均无明显异常;WHIM 综合征患儿的淋巴细胞免疫表型为 CD 3+、CD 19+细胞减少;PI 3 Kδ 过度活化综合征患儿CD19+细胞减少。5例患儿行鼻呼出气一氧化氮检测(fractional exhaled nitric oxide,FeNO),结果显示4例PCD患儿的中位FeNO值为30ppb(最小值4ppb,最大值213ppb),1例CF的FeNO值为156ppb。10例PCD 患儿接受了纤毛电镜检查,2 例纤毛结构未见异常,2例未见纤毛细胞,4例表现为纤毛数量减少,2例表现为微管排列不规则、内外动力臂缺失。
2.5 基因检查结果对临床诊断及治疗的影响
97例患儿中肺实质病变组89例,基因诊断前临床诊断主要考虑PID、PCD 和CF,最后通过基因确诊PCD20例、CF4例、PID2例、NMD2例,诊断率为31.5%。肺间质病变组8例,基因诊断前临床诊断广泛肺间质病变(原因不清),基因检测确诊为肺表面活性物质代谢障碍2型3例,诊断率37.5%。1例PID(WHIM综合征)患儿经基因检测确诊后转诊至免疫专科继续治疗、随访,1 例APDS、2 例NMD、3 例肺表面活性物质代谢障碍患儿因病情重、预后差、家庭经济情况等因素放弃治疗出院。
3 讨论
儿童疑难罕见病多是遗传性疾病,其中单基因病又被称为孟德尔遗传病,由明确的单个基因变异而引起临床表现。呼吸系统单基因遗传病种类多,包括肺部原发性疾病以及有肺部受累的其他遗传病。由于其较少被关注且临床表型缺乏特异性,容易漏诊。近年来,分子生物学尤其是二代基因测序技术发展迅速,测序成本不断降低,许多呼吸系统疑难病的诊断取得了突破性进展。有研究表明,对于临床表型复杂的疑难病,采用WES的诊断阳性率可达28.8%[2]。本研究97例患儿的总体诊断率为32.0%,与以往研究类似[3]。诊断疾病谱包括:PCD 20 例(64.5%),CF4例(12.9%),肺表面活性物质代谢障碍 3例(9.7%),PID 2例(6.5%),NMD 2例(6.5%)。通过NGS 测序本组确诊病例中原发于肺部的单基因遗传病以PCD最多见,结合北京儿童医院研究结果[3],推测在中国儿童群体中,PCD可能是最常见的原发于肺部的单基因遗传病。在本研究中,有66例基因检测结果为阴性,进一步分析这些病例,发现有部分检测出单个相关的异常基因。分析原因可能是受限于现有的检测手段,或存在未被证实的基因遗传模式。
过去,透射电子显微镜下典型的超微结构缺陷为诊断PCD 的金标准,然而近年来研究发现约1/3 PCD 患者纤毛超微结构正常[4]。本研究完善纤毛电镜检查的患儿也有1/5 未见异常,说明仅依靠电镜检查会漏诊部分患者。纤毛的超微结构改变可能是PCD特有,也可能与呼吸道感染或暴露于环境污染物中的获得性变化部分重叠[5]。而鼻FeNO、高速视频显微镜分析、针对纤毛蛋白的免疫荧光检测等灵敏度和特异度有限,复杂且价格昂贵,未在临床中广泛开展[6-7],因此基因诊断在PCD 中有不可或缺的作用。同时,区别于国外某些近亲结婚率高的地区致病变异较单一[8],国内PCD 患者具有较显著的基因异质性和基因变异异质性,因此在选择基因检测时仍应以WES 为主。在WES 未找到令人信服的致病变异的情况下,则可以选择进一步针对PCD 相关候选基因的拷贝数变异靶向分析或低深度的全基因重组测序分析可能发生于深度内含子的变异[9]。
CF 在国内较为罕见,尚无发病率的相关数据,仅有散发病例报道[10],且国内CF 患者以呼吸系统受累为主,消化系统症状轻微,仅凭临床表现及常规的辅助检查难以与PCD 区分。国内汗液试验开展较少,基因检测安全易行,对于中国CF 患者具有更重要的意义。针对CF的基因检测,国外多采用对CFTR基因已知的常见特异性变异进行检测[11]。我国关于CFTR变异的报道多为少见变异、变异范围广泛,具有遗传多样性,可选择参照标准序列进行CFTR全基因测序,有利于检测少见变异,甚至发现新的致病性变异。
以肺表面活性物质代谢障碍为代表的则是一组异质性的以肺间质受累为主的疾病,临床主要表现为气促、发绀,目前发现的主要变异基因有SFTPA 1、SFTPA 2、SFTPB、SFTPC、ABCA 3和NKX2-1[12]。虽然这类疾病的诊断主要依靠临床表现、影像学检查结合纤维支气管镜或肺活检的病理检查结果,但在以气促、弥漫性肺部间质性改变为主要表现的疾病,病因多且复杂,基因检测可快速作出诊断,避免有创操作及其他检查花费。
尽管目前对遗传性疾病治疗的研究有限,缺乏对因治疗的方法,但在分子层面确诊对不同基因型致病的机制、基因变异与特定信号通路联系的探索至关重要,也为生物制剂和小分子调节剂靶向治疗提供依据,进而实现精准医疗。针对CF、PID某些特定位点变异已有针对基因的治疗[13-14]。此外,基因检测不仅可明确诊断、指导治疗,还可扩展到对受影响的个人及其家庭成员进行适当的遗传咨询。
基因检测虽然在儿童呼吸系统疑难疾病的诊断中具有较大优势,但在实际应用中,仍存在明显的局限性。首先使用有针对性的基因测序包可减少脱靶,但由于部分疾病的遗传异质性高(如PCD、PID),可能导致漏诊。研究表明,目前分子诊断在PID 及PCD的阳性率仅50.0%左右[15-16]。其次,NGS允许更多的基因被平行测序,测试的敏感性增加,但其特异性降低。每个个体中存在的良性遗传变异的数量增加使得过度诊断的可能性很大,且随着测试规模的增加而增加。此外,目前商业化的NGS 多为外显子测序及目标基因包检测,无法通过测序发现深度内含子等部位的变异。部分疾病致病机制可能为位于不同基因上的变异构成的双基因遗传模式,进一步增加了基因诊断的难度。最后,NGS 技术价格昂贵、检测时间长、解释结果并非绝对可靠、许多基因疾病并无特异性治疗手段,无法在临床大规模推广。此外,本研究还存在其他局限性,为单中心、回顾性研究,病例数较少,患儿及家属可能存在回忆偏倚。
总之,由于临床和遗传的高度异质性、缺乏简单而广泛可用的辅助检查,儿童呼吸系统遗传性疾病的诊断具有挑战性。临床医师应不断提高对儿童呼吸系统遗传性疾病的认识,当对表型认识更全面、深刻时,有针对性地选择目标基因测序,可减少患者花费、降低社会医疗负担。对于一些不明意义的变异,未来仍需进行更多的功能研究。对不同变异进行正确的功能描述有利于实现精准医疗,早期诊断、早期纳入慢性肺部疾病管理并制定个体化治疗方案对延缓病程进展、改善部分患者的预后具有重要意义。
